El blog de la limpieza, desinfección y esterilizacion de dispositivos sanitarios. Este Blog no pertenece ni representa a ninguna Sociedad Científica, Asociación u Organismo, su finalidad es la difusión de conocimientos y actividades relacionados con la Esterilización. Todo es fruto de una búsqueda personal de evidencia en este campo sanitario. El administrador de este blog no se responsibiliza de la información contenida en el blog pues pudieran existir errores de intepretación o traducción en algún caso de los artículos o fuentes originales. Se recomienda, por tanto, consultar con los escritos originales (enlaces), de los que tampoco este administrador se responsabiliza de su exactitud. Tampoco se responsabiliza de las opiniones vertidas por sus seguidores. Los contenidos patrocinados se indicarán debidamente.
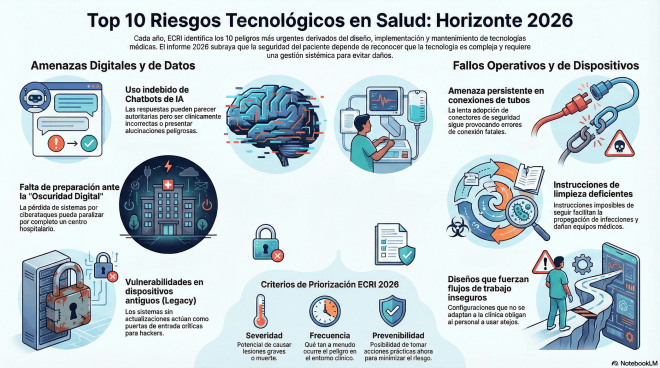
Este año el Top 10 de ECRI tiene mucho que ver con la IA, es el dilema de la tecnología infalible.
Existe la creencia de que a mayor sofisticación de las herramientas, menor es el margen de error. Sin embargo, la realidad que nos presenta ECRI tras 19 años de análisis es opuesta: a medida que la tecnología avanza, surgen vulnerabilidades invisibles y sistémicas. La seguridad del paciente no es una propiedad intrínseca de un dispositivo, sino el resultado de un «sistema total». Este equilibrio depende de la interacción crítica entre las personas, los procesos y el entorno. El informe de Riesgos Tecnológicos para 2026 nos advierte que el peligro no suele residir en un chip defectuoso, sino en la fragilidad de un sistema que confía ciegamente en infraestructuras que no comprende o que no puede controlar. Veamos unos ejemplos del Top 10.
¡Cuando los chatbots «alucinan» consejos médicos! El riesgo número uno para 2026 no es un fallo mecánico, sino una crisis de confianza algorítmica. El uso de Modelos de Lenguaje Extensos (LLM) como ChatGPT, Claude, Copilot o Grok se ha infiltrado en la clínica sin una regulación específica para el entorno sanitario. Estas herramientas no están diseñadas para la medicina; son motores de predicción estadística, no de comprensión clínica. El mayor peligro reside en las «alucinaciones» y en la predisposición de estos modelos a «complacer» al usuario. Ante una duda médica compleja, la IA prefiere ofrecer una respuesta fluida y autoritaria, aunque sea incorrecta o sesgada, antes que admitir su ignorancia. Esta búsqueda de la satisfacción del usuario, en lugar de la precisión clínica, convierte a la IA en un asesor de salud extremadamente poco fiable.
«El apagón electrónico». El colapso sistémico o schock que nadie espera. La dependencia absoluta de los sistemas electrónicos ha creado un flanco de ataque masivo. La «Oscuridad Digital» (una pérdida repentina de acceso a la información por ciberataques, fallos internos o desastres) ya no es una hipótesis de ciencia ficción; es una eventualidad operativa inminente. Y es un riesgo para los pacientes (decisiones de tratamiento a ciegas, sin acceso a historiales o resultados de laboratorio), clínicos (el personal se ve obligado a retomar flujos de trabajo manuales y obsoletos que no han practicado en años, operando bajo una presión cognitiva insoportable) y las organizaciones, con un caos.
El «Caballo de Troya» (dispositivos médicos heredados «Legacy»). Los dispositivos legacy (equipos antiguos que aún funcionan pero cuyo software ya no recibe parches de seguridad) son el «Caballo de Troya» moderno (Riesgo #8). La realidad económica impide a los hospitales renovar todo su parque tecnológico, dejando miles de puertas abiertas a los hackers. Un equipo de rayos X o un monitor de signos vitales comprometido no es solo un equipo fallido; es el puente que permite a un atacante saltar hacia el núcleo de la red hospitalaria. La solución para 2026 no es el reemplazo total (financieramente inviable) sino la segmentación agresiva de redes y la implementación de controles de seguridad compensatorios que aíslen estos puntos vulnerables de los sistemas críticos.
El desafío de los productos falsificados. El panorama geopolítico está alterando la seguridad del paciente. La retirada de apoyos internacionales (como la de EE. UU. de la OMS) y la reducción de personal en agencias de vigilancia sanitaria (Riesgo #3) han dejado un vacío de control. Esta falta de vigilancia externa delega en los equipos de compras la carga de detectar productos subestándar o falsificados.
¿Qué hay de lo mío? ¿Y sobre nuestro terreno? ¿Dice algo el informe? Pues si, en dos puntos, y los he tratado en el Blog previniendo de su importancia. Este año no aparecen los endoscopios ni duodenoscopios, pero lo que hay son cosas muy simples pero que son la base de nuestro trabajo.
7. Instrucciones de limpieza deficientes: Manuales de fabricantes incompletos o imposibles de seguir que facilitan la propagación de infecciones. Son las IFUs y la necesidad de que tengamos toda la información en las centrales, y por supuesto la importancia de la limpieza.
Una IFU no es un lujo o accesorio es OBLIGATORIO.
10. Problemas de calidad del agua en esterilización: El agua impura puede dejar residuos infecciosos o dañar el instrumental quirúrgico. ¿Cuántas veces he hablado del agua en el Blog? ¿Y el circulo de Sinner? ¿Y de la mezcla agua y detergentes?
Esta duda se planteó en el II Curso de Reprocesado de Instrumental Médico realizado en Oviedo del 6 al 8 de noviembre de 2005. Gracias a Jorge y Mamen por la excelente organización. Este curso tiene mucho futuro.
Sé que me estoy metiendo en un buen «freago», pero es que es un campo en continuo desarrollo y tenemos que ir aclarando conceptos. Además, esta es la entrada 300 del Blog y hay que meterse en temas controvertidos.
El RD 192/2023 regula los productos sanitarios de uso humano en España, incluyendo sus accesorios, y hará de acompañamiento al Reglamento (UE) 2017/745 (“MDR”) que es de aplicación directa en la UE.
Si un hospital recibe un implante de tipo IIb o III (alto riesgo, como prótesis cardíacas, implantes neurológicos, implantes óseos, dispositivos vasculares, entre otros), y el fabricante proporciona en las IFU (Instrucciones de Uso o para el usuario final)procedimientos específicos de limpieza, reprocesamiento y esterilización, es fundamental cumplir con una serie de consideraciones técnicas y regulatorias obligatorias:
1. Validación del proceso por parte del fabricante
El proceso descrito en la IFU debe haber sido validado por el fabricante. Esto implica que el fabricante ha demostrado, mediante estudios documentados, que el reprocesamiento no compromete la seguridad, la funcionalidad ni la biocompatibilidad del dispositivo. El hospital solo podrá realizar la limpieza o esterilización conforme a esas instrucciones validadas.
Su uso está previsto una sola vez (single use), salvo indicación contraria explícita y validada por el fabricante. Y que quiere decir ésto, pues que si se te infecta o se complica la cosa, no vale con quitarlo y que nosotros lo reprocesemos otra vez (OJO CON ÉSTO).
Y aquí pregunto: ¿tenemos validadas nuestras lavadoras termodesinfectadoras? ¿y nuestros autoclaves y esterilizadores? Pues, en general, NO. Entonces, ¿podemos asegurar al usuario final que el producto final que le entregamos va a ser como él lo espera? Si en al menos uno de los dos casos tu respuesta ha sido NO, entonces no te metas en este lío. Otro OJO, validar no es poner indicadores biológicos. Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.
2. Cumplimiento estricto de las instrucciones
El establecimiento sanitario debe seguir de manera exacta todos los pasos indicados por el fabricante: tipo de detergente, método y parámetros de esterilización (temperatura, presión, tiempo, tipo de ciclo), tipo de envase o empaque, controles de esterilidad, etc. Cualquier desviación invalida la garantía del proceso y traslada la responsabilidad al hospital.
El fabricante ha validado el proceso de reprocesamiento, incluyendo limpieza, desinfección y esterilización, y lo describe en la IFU.
Tú (el hospital) sigues exactamente los pasos, parámetros y materiales especificados (p. ej., tipo de detergente, temperatura, método de esterilización).
El fabricante asume la responsabilidad de que el dispositivo sigue siendo seguro y eficaz después del proceso, siempre que tú sigas sus instrucciones al pie de la letra.
👉 En este caso, el hospital no se convierte en “fabricante” ni asume responsabilidad adicional, ya que está actuando conforme al uso previsto y validado por el fabricante.
3. Responsabilidad del fabricante y del hospital
Mientras el hospital actúe conforme a las IFU, la responsabilidad de la seguridad del dispositivo tras el reprocesamiento recae en el fabricante. En cambio, si el hospital modifica el proceso, utiliza métodos no validados o reprocesa un dispositivo declarado de “un solo uso”, entonces asume el rol de fabricante según la legislación aplicable (p. ej., Reglamento (UE) 2017/745, artículo 17), con todas las obligaciones regulatorias asociadas.
Para productos implantables, el RD regula que junto al producto se debe entregar al paciente la “tarjeta de implante” que contenga información (por ejemplo: identificación del producto, número de lote, etc.). También se establecen obligaciones de registro nacional de implantes para que los centros y profesionales comuniquen datos.
4. Reprocesamiento de dispositivos de un solo uso
Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.
Todo procedimiento de limpieza y esterilización debe estar documentado, trazable y controlado dentro del sistema de gestión de calidad del hospital. Esto incluye:
Registro de lote o número de serie del dispositivo.
Identificación del personal responsable.
Validación y control de los equipos de reprocesamiento.
Verificación de los parámetros del ciclo de esterilización.
Situación
¿Se puede limpiar/esterilizar en el hospital?
Condición
IFU incluye instrucciones validadas por el fabricante
✅ Sí
Siguiendo exactamente las instrucciones validadas
IFU indica “single use only” o no describe reprocesamiento
❌ No
Sería un reprocesamiento no autorizado → el hospital pasa a ser “fabricante”.
Implante de tipo III sin validación del proceso
❌ No
Riesgo alto, no permitido sin validación y certificación específica.
La entrada publicada el 06/01/2026 está teniendo cierto eco, y he recibido un comentario de Xavier Canals de Tecnomed Ingenieros Consultores que añade información que puede ser útil para todos y copio textual en cursiva:
Solo unas consideraciones regulatorias…
«1. Validación del proceso por parte del fabricante»; si no estuviera validado, no le habrían dado el marcado CE. En caso de duda sobre el proceso, solicitar información adicional al fabricante. Un único caso donde podríamos no tener esta información es en implantes que ya vienen estériles (lo más normal) o en los implantes a medida donde el fabricante es el propio hospital, por ejemplo. (hay que solicitarselo o … no usarlo.
» Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.» Entiendo que la frase correcta es «y que puede no coincidir con la empresa fabricante del equipo»,
en general, el fabricante es el que tiene más facilidad de realizar la validación y, en su caso, reparar el equipo si no pasa la validación Debe incluir también una parte de la validación a nuestro servicio, procedimientos y registros… Imaginaros en un símil doméstico alguien que lavara prendas de color en el programa de la lavadora de algodón de 90 ºC… La lavadora, ok, pero el mal uso produce resultados defectuosos.
«4. Reprocesamiento de dispositivos de un solo uso Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.» El reglamento MDR indica que, al no llegar a un acuerdo de todos los países europeos, cada país debía establecer su autorización. Para España, el RD 192/2023 establece que sí se pueden reprocesar productos sanitarios de un solo uso, pero que precisan licencia, segregando el caso de «fabricante de producto reprocesado» (art.12) del de «reprocesamiento en hospitales» (art.13) y sus subcontratistas «reprocesadores externos» (art.14)
Ahora solo está permitido el de los «fabricantes de producto reprocesado», y que no tengo conocimiento de que haya ninguno autorizado. Para los hospitales, según la disposición final tercera: » 2. Las actividades de reprocesamiento de productos de un solo uso en hospitales establecidas en el capítulo III, (incluida la subcontratación de estas actividades a un reprocesador externo) requerirán el previo desarrollo por el Ministerio de Sanidad de los requisitos técnicos establecidos en este real decreto», es decir, que hay que esperar a un decreto nuevo.
y hay que tener en cuenta que el RD 192/2023: «Artículo 15. Utilización de productos de un solo uso reprocesados. … 2. No se permitirá la adquisición y utilización en España de productos que hayan sido transferidos a un tercer país para su reprocesamiento…»
Colofón final, y respuesta a la pregunta de la entrada:
¿Es posible hacerlo legalmente en España? SI
¿Es probable que tengamos todos los elementos necesarios para hacerlo? NO
Conclusión: NO, al menos de momento.
Deseo que en el día de los Reyes Magos hayáis recibido muchos regalos y no estéis leyendo esta entrada en el Blog porque os han traído la corbata o los calcetines de todos los años. Yo tengo un nuevo estuche para mi trompeta.
Os espero en el próximo concierto de año nuevoQué Melchor más guapo!
Espero ayudar en lo que pueda con esta entrada. Se puede liar la cosa como el que montan «El canijo de Jerez» y «Los Estanques».
Sé que las entradas que tienen que ver con reglamentación, normativas… son muy aburridas y tediosas. Pero es que hay que conocer la legislación vigente sobre productos sanitarios. El trabajo de las centrales de esterilización va más allá de poner un Bowie o de controles.
El Reglamento 2017/745 sobre dispositivos o productos sanitarios habla de los IFUs o instrucciones de uso (dicho en español) y de su importancia en la calidad y seguridad del paciente. En la Guía de la AEMPS nos dice que la IFUs es la información facilitada por el fabricante para informar al usuario sobre la finalidad prevista de un producto, su uso correcto y las precauciones que deban tomarse (apartado 14 del artículo 2). En esta IFU deben aparecer los medios para limpiar, desinfectar y esterilizar el material, ya que ahora mismo las supervisoras de las centrales de esterilización deben estar pidiendo y casi rogando esta información a las casas comerciales.
Todos los productos deben ir acompañados de la información sobre seguridad y funcionamiento necesaria para utilizarlos de forma segura y que ayude a identificar tanto el producto como el fabricante y/o el representante autorizado, siempre teniendo en cuenta la formación y los conocimientos de los posibles usuarios. Esta información incluye la etiqueta, el embalaje del producto y los datos en las instrucciones de uso. Como excepción a los principios generales, no se requieren instrucciones de uso para los productos de Clase I si pueden utilizarse de forma adecuada sin dichas instrucciones. Lo más probable es que se presente una excepción para los productos de Clase Ir, ya que el reprocesamiento (limpieza y esterilización) sí requerirá instrucciones. Los requisitos relativos a la información que debe suministrarse con el producto se encuentran en el anexo I, apartado 23 del capítulo III y artículo 103 del Reglamento (UE) 2017/745. En el etiquetado y las instrucciones de uso, así como en los materiales promocionales del producto, el fabricante no puede (artículo 7):
Atribuir funciones y propiedades que no tenga el producto.
Crear una impresión falsa con respecto al tratamiento o diagnóstico, funciones o propiedades que el producto no tenga.
No informar al usuario o al paciente de un riesgo probable asociado con el uso del producto en línea conforme con su finalidad prevista.
Sugerir usos para el producto distintos a los que, según el MDR, formen parte de la finalidad prevista para la que se llevó a cabo el procedimiento de evaluación de la conformidad.
El marcado CE deberá figurar también en las instrucciones de uso, así como en cualquier envase de venta.
Está prohibido poner marcas que puedan inducir a error a terceros en relación con el significado del marcado CE. Se pueden colocar otras marcas adicionales en el producto, el embalaje o las instrucciones de uso, pero estas no podrán afectar negativamente a la visibilidad o la legibilidad del marcado CE.
Especialmente importante es el contenido referente a los avisos y advertencias. En realidad, es esencial que los avisos y advertencias que el fabricante considera necesarios comunicar al usuario, estén reflejados en él. Un proceso eficaz de gestión de riesgos debe aplicar las medidas consideradas necesarias. Por orden de prioridad, primero lo harán las relacionadas con el diseño del producto sanitario. Después, las que aplican cambios en los procesos relacionados con el producto sanitario. Tras ellas, está la información para la seguridad. Esta información es el propio contenido del IFU, junto con los avisos y advertencias (warnings) que el propio fabricante considere necesarios.
Las instrucciones de uso de un producto sanitario (IFU) deben contener al menos los siguientes datos, según el producto del que se esté tratando (tomado de https://productosanitario.es/):
Denominación o nombre comercial del producto.
Nombre, nombre comercial registrado o la marca registrada del fabricante y su domicilio social.
Marcado CE del producto.
Si se trata de un producto de un solo uso o es un producto reutilizable.
Finalidad prevista del producto, indicando sus indicaciones y beneficios clínicos esperados, contraindicaciones, riesgos residuales y efectos secundarios indeseables, así como las medidas de precaución apropiadas y el grupo de pacientes al que está indicado el producto.
Forma de eliminación segura, tanto del producto como de cualquier sustancia residual.
Si el producto se suministra estéril, una indicación de su estado estéril y el método de esterilización.
Características del funcionamiento del producto.
Especificaciones que necesita el usuario para utilizar el producto de forma adecuada.
Datos sobre la preparación o manipulación del producto antes de ser utilizado.
Datos de mantenimiento, limpieza, conservación y cambio de consumibles si es necesario.
Fecha de publicación de las instrucciones de uso o, si han sido revisadas, fecha de publicación e identificador de la última revisión.
Un aviso destinado al usuario o paciente de que cualquier incidente grave relacionado con el producto debe comunicarse al fabricante y a la autoridad competente del Estado miembro en el que estén establecidos el usuario y/o el paciente.
En las instrucciones de uso estará prohibida la utilización de textos, denominaciones, marcas comerciales, fotografías e imágenes u otros signos que puedan inducir a error al usuario o al paciente en cuanto a la finalidad prevista, la seguridad y el funcionamiento del producto. El fabricante debe de tener especial cuidado al redactar las instrucciones de uso para no cometer los errores de dar a entender usos del producto diferentes a su finalidad prevista, atribuir al producto funciones y/o propiedades que no posee, crear una falsa impresión sobre tratamiento o diagnóstico, funciones o propiedades que el producto no posee y no informar al usuario o al paciente sobre los posibles riesgos que conlleva la utilización del producto conforme a su finalidad prevista.
En un estudio de APIC mediante encuesta a 1.198 preventivistas o secciones de control de infecciones, mostró que el 42% de sus centros habían sido citados (por temas legales) por no respetar una IFU, el 84% de los preventivistas tuvieron que ponerse en contacto con un fabricante para aclarar la limpieza, desinfección o esterilización adecuadas de un producto, y el 8% dieron el paso adicional de ponerse en contacto con la FDA para obtener aclaraciones sobre las instrucciones de uso.
La APIC considera que se trata de una carga inaceptable que no apoya el objetivo de prevenir la transmisión de las HAI (IRAS). La APIC pide lo siguiente:
El desarrollo de herramientas para ayudar al personal sanitario en los procesos de limpieza, desinfección y esterilización de instrumentos médicos.
Informar a los fabricantes y a la FDA sobre las IFU problemáticas.
Educar a los responsables políticos y a las organizaciones sanitarias sobre las deficiencias del marco normativo actual que limitan la capacidad de los PI para proteger a los pacientes de la transmisión de las HAI a través de dispositivos médicos.
Convocar a las organizaciones interesadas a trabajar con APIC para proponer un nuevo marco regulatorio para la limpieza, desinfección y esterilización de dispositivos médicos.
Un repositorio público de instrucciones de uso para que los usuarios tengan acceso a la información adecuada sobre los productos que ya no se fabrican y/o cuando el fabricante ya no esté en activo.
En este enlace tenéis el Informe de APIC, que es bastante extenso (está en inglés).














Debe estar conectado para enviar un comentario.