El blog de la limpieza, desinfección y esterilizacion de dispositivos sanitarios. Este Blog no pertenece ni representa a ninguna Sociedad Científica, Asociación u Organismo, su finalidad es la difusión de conocimientos y actividades relacionados con la Esterilización. Todo es fruto de una búsqueda personal de evidencia en este campo sanitario. El administrador de este blog no se responsibiliza de la información contenida en el blog pues pudieran existir errores de intepretación o traducción en algún caso de los artículos o fuentes originales. Se recomienda, por tanto, consultar con los escritos originales (enlaces), de los que tampoco este administrador se responsabiliza de su exactitud. Tampoco se responsabiliza de las opiniones vertidas por sus seguidores. Los contenidos patrocinados se indicarán debidamente.
Ya sabéis que soy bastante crítico con las certificaciones UNE-EN ISO o cualquier otro sistema similar. Sería largo de explicar, pero aquí va una pincelada.
Pregunta: ¿Disponer de una certificación UNE EN ISO 9001 o de la UNE EN ISO 13485 de productos sanitarios tiene alguna validez legal? ¿O te da un respaldo legal que te ayude en caso de problemas de responsabilidad en una central de esterilización de un hospital? Este asunto salió en el II Curso de Reprocesado de Instrumental Médico realizado en Oviedo del 6 al 8 de noviembre de 2005.
🔹 1. Naturaleza de las certificaciones ISO
El Real Decreto 192/2023 regula los productos sanitarios de uso humano en España, incluyendo sus accesorios, y hará de acompañamiento al Reglamento (UE) 2017/745 (“MDR”) que es de aplicación directa en la UE.
Las normas UNE-EN ISO 9001 (gestión de calidad) y UNE-EN ISO 13485 (sistemas de gestión de calidad para productos sanitarios) son normas voluntarias, no leyes. Esto significa que no tienen validez legal directa, en el sentido de que no sustituyen ni eximen del cumplimiento de la legislación sanitaria o de responsabilidad civil o penal. Y es que nos certificamos de lo que queremos, es el famoso ALCANCE, quiere decir que puedo limitar la certificación a una pequeña parte de mis procesos, y el auditor certificará que es cierto que lo hago. Puedo decir que todos mis contenedores van a salir de la central con una margarita. ¿Sirve de algo? No, pero me puedo certificar.
Sin embargo, sí tienen valor jurídico indirecto o probatorio. Vamos, que te pueden ayudar ante un juez o un problema legal.
🔹 2. Validez legal directa
Ninguna de las dos certificaciones otorga por sí misma licencia, autorización o habilitación legal para operar una central de esterilización ni para fabricar o manipular productos sanitarios. Quien lo da es el RD 192/2023.
La UNE-EN ISO 13485 puede ser requisito técnico exigido por las autoridades sanitarias o por clientes en algunos procesos de certificación de productos o contratos públicos, pero no sustituye la conformidad reglamentaria (por ejemplo, el marcado CE o las normas de higiene y trazabilidad hospitalaria).
🔹 3. Valor como respaldo legal o en el caso de problemas.
Aunque no te exime de responsabilidad, sí puede servir como elemento de defensa o atenuante en procedimientos legales o administrativos:
Demuestra diligencia y cumplimiento de estándares reconocidos internacionalmente.
Puede ayudar a probar que se aplicaron medidas razonables de control y gestión de calidad.
En un proceso judicial por una supuesta negligencia o fallo en la esterilización, tener un sistema certificado ISO 9001 o ISO 13485 puede servir como evidencia documental de que el centro tenía procedimientos controlados, auditados y validados.
👉 Es decir, no te “protege” legalmente de una demanda o sanción, pero sí mejora tu posición defensiva al mostrar diligencia debida y gestión responsable.
🔹 4. En el contexto de una central de esterilización hospitalaria
El cumplimiento de ISO 9001 o 13485 puede ayudar a garantizar trazabilidad, validación de procesos y control documental, que son claves para la seguridad del paciente.
Además, puede facilitar auditorías sanitarias o acreditaciones hospitalarias (como Joint Commission, ACSA, etc.).
Pero, ante un incidente, la responsabilidad recaería igualmente en los profesionales o la institución si se demuestra una mala praxis o incumplimiento de la normativa sanitaria.
🔹 5. Resumen
Aspecto
ISO 9001 / ISO 13485
Valor legal
Obligatoriedad
Voluntaria
No sustituye requisitos legales.
Licencia o autorización
No
Ninguna.
Valor probatorio
Sí
Sirve como evidencia de buena práctica.
Protección ante sanciones o demandas
Parcial / indirecta
Puede mitigar o apoyar la defensa.
Relevancia práctica
Alta (mejora procesos y seguridad)
Importante en auditorías o litigios.
No he localizado ninguna sentencia que establezca de forma general y vinculante que la certificación ISO 9001 o ISO 13485 exime de responsabilidad civil o penal en el ámbito sanitario. En la práctica judicial sobre responsabilidad sanitaria (negligencias, mala praxis, responsabilidad patrimonial) los tribunales valoran un conjunto de pruebas: historia clínica, protocolos, formación del personal, mantenimiento/validación de equipos, registros de procesos y, entre ellos, la existencia de sistemas de gestión o certificaciones. Una ISO puede fortalecer la defensa (demostrar diligencia y controles), pero raramente será decisiva por sí sola. Varias resoluciones y sentencias (contencioso-administrativas) reconocen que un certificado ISO 9001 puede ser un criterio válido o un medio para acreditar solvencia técnica en concursos públicos; es doctrina consolidada en materia de contratación administrativa que no obliga a excluir otros medios alternativos de acreditación. Esto muestra valor administrativo/probatorio de las ISO, pero en un contexto distinto al de la responsabilidad sanitaria.
Según la web de unos juristas dedicados a negligencias médicas (descubrí esta página en el curso con Mamen):
Disponer de una certificación UNE-EN ISO 9001 o ISO 13485 no te da inmunidad legal, pero sí constituye una prueba sólida de que la organización aplica buenas prácticas de gestión y control de calidad reconocidas internacionalmente, lo que puede ser muy útil como respaldo legal o reputacional en caso de inspecciones, auditorías o litigios.
Se me va el santo al cielo con estos temas, como Sonora Dinamita que se le perdió la cadenita a Carmen. ¡¡Cómo me gustan esas trompetas al fondo!! Se me van los pies y los dedos, y eso que solo tocan tres notas.
Para que no digáis que pongo música freaky, más de uno y una no se esperaba alos Cure en este blog:
No a la guerra. Nuestro apoyo a los ciudadanos de Ucrania
«La que se avecina» es el título de la ponencia sobre el nuevo Reglamento, pero la geopolítica se ha colado y también podemos aplicarlo.
Hace unos días «tomé un café» en una sesión organizada por la casa comercial Dr. Weigert sobre el Reglamento 2017/745 (link de la noticia), del que ya hablé en el Blog en una entrada, pero esta vez lo voy a ampliar.
Lugar del encuentro
Me acompañó una amiga y profesional como Mercedes García Haro, que se centró en la aplicación directa en la RUMED. Fue de esas sesiones con amigos, en un ambiente muy distendido y agradable. Una organización inmejorable con el equipo de Dr. Weigert.
Aquí Mercedes iniciando la exposición
¿Qué debo hacer con el nuevo reglamento de productos sanitarios? (así se llamaba la charla). Leerlo y padecerlo en la intimidad como unas hemorroides.
Y aquí empezando mi charla
Este reglamento tiene como objetivo garantizar la disponibilidad en el mercado de productos sanitarios eficaces, de calidad y seguros.
De dónde venimos, y adonde vamos
Reglamento de Productos Sanitarios o Medical Devices Regulation (MDR), son dos conceptos: Reglamento y Productos sanitarios (Artículo 2 del Reglamento 745/2017).
Los reglamentos son actos jurídicos que se aplican de manera automática y uniforme en todos los países de la UE desde su entrada en vigor, sin necesidad de incorporación al derecho nacional.
Son obligatorios, en todos sus elementos, en los Estados miembros. El Reglamento comunitario una norma de aplicación directa, el RD 1591/2009 ha pasado a quedar, en aquellos aspectos que no resulten conformes con aquel, desplazados (que no derogados). El Reglamento es una norma de aplicación directa, pero se supedita algunos aspectos o cuestiones a la regulación que se establezca a nivel nacional. El Reglamento obliga a:
1.- Derogar el Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios (excepto 21, 38, 39 y 40, y el Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos (excepto 18, 34, 35 y 36), ante la aplicación directa del Reglamento (UE) 2017/745.
2.- Desarrollar las medidas reglamentarias necesarias para aquellos aspectos en los que el reglamento comunitario ha determinado que serán los Estados miembros lo que establecerán la regulación a nivel nacional.
3.- Adaptar, adoptar o mantener las medidas requeridas por la legislación nacional.
El futuro Real Decreto es necesario para establecer:
a) Los requisitos y procedimientos para la regulación de los productos fabricados y utilizados en un centro sanitario (fabricación in house)
b) Los requisitos y procedimientos para la regulación del reprocesamiento de productos sanitarios de un solo uso
c) La regulación de la tarjeta de implantación
d) La creación de un registro nacional de comercialización de productos sanitarios
e) Establecer que la autoridad competente es la Agencia Española de Medicamentos y Productos Sanitarios independientemente de las competencias de otras autoridades sanitarias
Según el proyecto de Real Decreto: Todas las centrales de esterilización o RUMED de los hospitales (públicos y privados) tendrán que obtener la Licencia de Funcionamiento.
¿Qué es un producto sanitario?Producto sanitario (Medical Devices: MD) es cualquier instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en seres humanos y que no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.
También se considerarán productos sanitarios los productos de control o apoyo a la concepción. Y atención, los productos destinados específicamente a la limpieza, desinfección y esterilización de los productos sanitarios. Se excluyen las lentes de contacto y productos para introducirse o colocarse en los ojos, y los productos invasivos quirúrgicos para modificar anatomía o fijación partes del cuerpo, excluidos tatuajes y piercings.
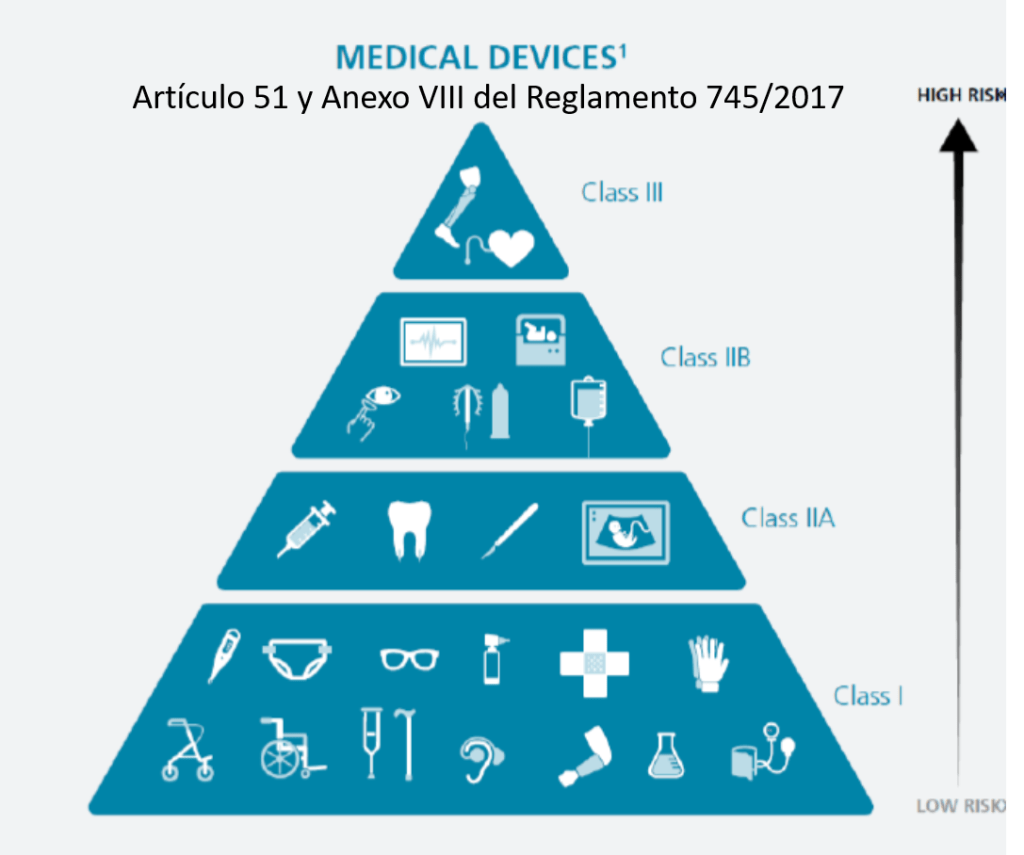
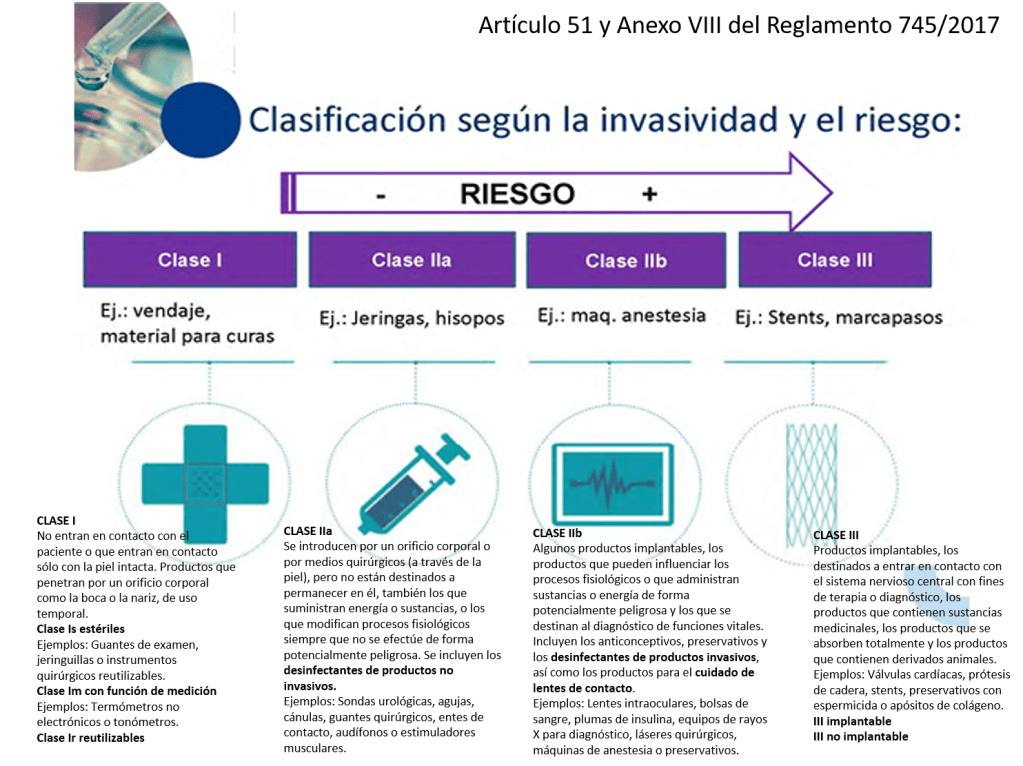
¿Cómo se clasifican los productos sanitarios? (Artículo 51 y Anexo VIII del Reglamento 745/2017)
Este reglamento tiene unos cambios relevantes que se aplican a la Central de Esterilización o RUMED:
•Los desinfectantes de productos no invasivos son IIa
•Los desinfectantes de productos invasivos son IIb
•Las lavadoras desinfectadoras pasan a ser productos sanitarios de la clase IIb
•Los equipos de esterilización pasan a ser productos sanitarios de la clase IIa
•Los indicadores químicos y biológicos NO tienen la consideración de productos sanitario (Confirmado por la AEMPS que se lo ha dicho a un amigo). La empresa GKE ha elaborado un documento donde dicen que «The GKE cleaning process monitoring indicators (CPI) are no medical devices in all countries and do not require any registration» (Párrafo actualizado 21/04/2022).
•Los productos fabricados por el propio hospital para uso interno (in house) pasan a estar regulados y precisan de documentación técnica y sistema de calidad ISO 13485 (¡¡no la 9001!!).
Aunque el Reglamento 2017/745 es de aplicación directa, hay determinados aspectos como el reprocesamiento, el régimen lingüístico o cuestiones de la fabricación en centros sanitarios, entre otros, que el reglamento determina que serán los Estados miembros los que establecerán la regulación a nivel nacional. Es por esto que en la actualidad se está elaborando el nuevo Real Decreto de Productos Sanitarios (sustituye al RD 1591/2009). Un producto sanitario:
-Se utiliza en personas.
–Finalidad prevista: Se diseña para cumplir un determinado fin. Documentada en el expediente técnico
-El fabricante lo ha validado en relación con el objetivo perseguido. Este objetivo lo establece el fabricante, y él mismo lo confirma, para emplearlo en relación con la enfermedad, discapacidad, proceso fisiológico, o patológico.
-Se ha diseñado para conseguir unos beneficios clínicos, siempre superiores a los posibles riesgos, y que él mismo ha sido capaz de evidenciar y justificar.
•Cuando se introduce un producto en el mercado, o lo pone en servicio, el fabricante se asegurará que se ha diseñado y fabricado con arreglo a los requisitos generales de producto sanitario.
•Realizar una evaluación clínica en relación con los requisitos esenciales de producto, incluyendo un seguimiento postcomercialización.
•Los de aquellos productos que no sean a medida, elaborarán y actualizarán la documentación técnica de dichos productos, permitiendo la evaluación de la conformidad de producto.
•Cumplir con las obligaciones de identificación y las de registro.
¿Cómo lo valida el fabricante? ¿Cómo el fabricante es capaz de determinar que, ciertamente, es capaz de ser eficaz y seguro para lo que él mismo pretendía Mediante el ejercicio de la evaluación clínica. La evaluación clínica es un proceso continuo, sistemático y planificado que pretende conseguir, evaluar y analizar información clínica. Estas normas pueden sernos de ayuda:
Y todo ello, en un sistema de seguridad y funcionamiento, que deberá disponer de un sistema de gestión de la calidad (UNE-EN ISO 13485 y la Guía o ayuda la UNE 179003:2009) y establecer, documentar, aplicar y mantener un sistema de gestión de riesgos (UNE-EN ISO 14971). Los estados miembros pueden permitir a los hospitales no aplicar todos los requisitos si la seguridad del nuevo producto son similares a las de los productos originales, el reprocesado se hace según especificaciones comunes sobre gestión de riesgos, la validación de procedimientos, liberación paramétrica o no de productos, ensayos, sistemas de calidad, etc… Solo se reprocesarán productos que se considere seguro hacerlo (por eso van a obligar a los hospitales obtener una Licencia de Funcionamiento). Los estados miembro pueden aplicar estas excepciones también a productos que se reprocesan en empresas externas a los hospital (PERO el producto vuelve al mismo hospital). Parece que los centros sanitarios y los fabricantes externos tendrán un régimen especial. Se abre la posibilidad a los «third party», aunque cada país puede imponer limitaciones al reprocesado y el uso de productos reprocesados. El reprocesamiento se lleva a cabo según especificaciones comunes sobre gestión de riesgos, validación de los procedimientos, liberación, sistema de gestión de la calidad, notificación de incidentes, trazabilidad.
En cuanto a los recursos humanos, se insiste en la figura del Responsable o Director Técnico, con unas funciones que ya conocemos:
•Supervisar las actividades de fabricación.
•Comprobar que se cumplen los requisitos exigidos por la reglamentación.
•Supervisar el archivo documental.
•Revisar y evaluar los incidentes.
•Ser interlocutor con las autoridades sanitarias y facilitar la documentación requerida.
•Solicitar la Licencia de Funcionamiento
Y aparece una nueva figura, que es el Responsable del cumplimiento del Reglamento (Artículo 15 Reglamento 745/2017), que tendrá esta formación, como Licenciado o Grado en Derecho, Medicina, Farmacia, Ingeniería u otra pertinente y un año experiencia en asuntos reglamentarios o SGC de productos sanitarios o cuatro de experiencia en asuntos reglamentarios o en SGC de productos sanitarios.
¿Y con todo esto ya vale?
Pues no, aun tenemos que (Artículo 10 del Reglamento 745/2017):
•Mantener a disposición de las Autoridades la documentación, la declaración de conformidad durante un periodo mínimo de 10 años desde la puesta en el mercado del último producto (15 años si son implantables).
•Disponer de un sistema de registro e información a las autoridades ante incidentes graves o acciones correctivas.
•A petición de la Autoridad competente, facilitarle toda la información para demostrar la conformidad de producto.
•En caso de que el fabricante encomiende a un tercero el diseño o fabricación del producto, se recogerá en la documentación.
•Las personas físicas o jurídicas podrán reclamar indemnizaciones por daños o perjuicios causados por un producto defectuoso; con arreglo al derecho nacional, o de la Unión, aplicable.
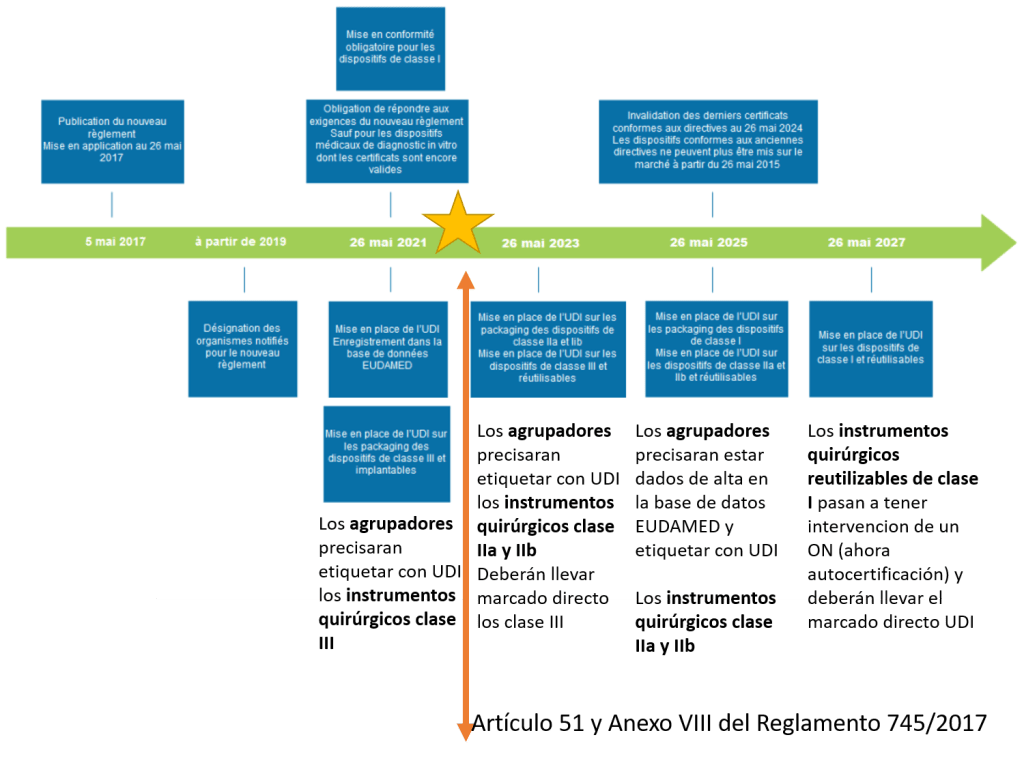
•Creación de una nueva base de datos llamada EUDAMED (acceso a las autoridades, la industria, los profesionales sanitarios y el público general) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
•Mejora en la trazabilidad de los productos sanitarios, mediante la implantación de un número de identificación único (UDI) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
Para terminar unas breves reflexiones en voz alta:
Todo este Reglamento puede ser un revulsivo para las RUMED, por el papel central en la seguridad del paciente que se les da, pero ¿estarán todas preparadas? Yo creo que no. Algunos hospitales o centros pequeños no tiene personal cualificado suficiente, maquinaria y equipos validados, presupuesto para desarrollarlo…
Creo que va a haber un boom de empresas especializadas en dar cobertura a todos los hospitales. Unos dando formación y asesoría en el Reglamento, sobre todo en los aspectos normativos, sistema de gestión de la calidad y sistema de gestión de riesgos.
Y otras empresas dando servicios mediante subcontratación o externalización de la RUMED o de la Dirección Técnica y Normativa.
No sabemos cómo será el futuro real decreto pero puede cambiar toda nuestra filosofía de trabajo.
Sé que es una entrada dura y con mucha información, y con muchas dudas que tendremos que ir solucionando.
El autor no tiene conflicto de intereses con la empresa Dr. Weigert
Y la despedida con uno de los mejores temas de los 70-80, con las Baccara y su «Yes Sir, I can Boogie» que se ha convertido en el himno no oficial de la selección escocesa de futbol, donde juega el petardo de Bale. En esos años, sólo sabían inglés Jesús Hermida y el Príncipe Gitano.
Alguno se sorprenderá por esta entrada, y dirá «otra vez».
No quiero que nadie se equivoque y voy a hacer un spoiler: Esta prohibido en España reesterilizar productos de un solo uso (y según creo), lo será por mucho tiempo.
Mientras tanto, la Comisión Europea está trabajando en unos requerimientos comunes para el reprocesamiento de productos sanitarios de un solo uso, en el ámbito del Reglamento 2017/745 (art. 17.3)
Si alguien quiere hacer alegaciones, tiene de plazo hasta el 20 de agosto para recibir comentarios al texto Borrador.
Personalmente me han llamado la atención estas cosas:
Art. 8.1: Hay que determinar el número máximo que se puede reprocesar un dispositivo
Art. 9.3: La documentación del proceso deberá custodiarse 10 años
Art. 10: Todos los procesos estarán validados (desde la limpieza hasta la esterilización)
Art. 10.8: Especifica que la monitorización deberá estar avalada o suplementada por indicadores biológicos (no dice cuáles)
Art. 11 al 19 son los diferentes pasos del proceso
Art. 20: Es la información que deberá aportar el fabricante o reprocesador al usuario
Art. 21: Habrá un sistema de calidad, pero no dice cuál (¿9001?, 13485, 179003)
Feliz lectura para la siesta de verano, ahora que soy padre comprendo a los míos cuando me mandaban callar o me ponían delante de la tele (la única) con «Verano azul», «Fama», «El coche fantástico», «El superhéroe americano», «Kung Fu». Esas tórridas tardes manchegas.
Sólo si ya no cumples los 40 años conocerás estas series.
Sólo teníamos las TV1 y la UHF, no había Youtube, Netflix, HBO… pero teníamos mucha imaginación adolescente.
Hoy entra en vigor el Reglamento de la Unión Europea 2017/745 sobre Productos Sanitarios y su fecha de aplicación según el periodo transitorio de 3 años, que se indica en su articulo 123, será el 26 de mayo de 2020.
El reprocesado y uso de productos reprocesados tiene que estar permitido por leyes nacionales. Así que de momento en España seguimos con el RD 1519/2009, y por eso sigue estando prohibido.
Los reprocesadores de productos de un solo uso se convierten en fabricantes, y deben asumir todas las responsabilidades y obligaciones que ello supone. De momento las centrales de los hospitales están todavía lejos de conseguirlo, ya que deben validar equipos (que es lo más costoso económicamente) y tener un sistema de gestión de la calidad y de gestión de riesgos. Además de la omnipresente UNE-EN ISO 9001, debemos cumplir otras normas (UNE-EN ISO 13485, UNE-EN ISO 14969 y la UNE-EN ISO 14971). Tenemos como guía o ayuda la UNE 179003:2009
Los estados miembros pueden permitir a los hospitales no aplicar todos los requisitos si:
La seguridad y rendimiento del nuevo producto son similares a las de los productos originales
El reprocesado se hace según especificaciones comunes sobre gestión de riesgos, la validación de procedimientos, liberación paramétrica o no de productos, ensayos, sistemas de calidad, etc…
Solo se reprocesarán productos que se considere seguro hacerlo
El cumplimiento de las especificaciones se certifica por un Organismo Notificado
Los estados miembro pueden aplicar estas excepciones también a productos que se reprocesan en empresas externas a los hospital (PERO el producto vuelve al mismo hospital). Parece que los centros sanitarios y los fabricantes externos tendrán un régimen especial
Se abre la posibilidad a los «third party», aunque cada país puede imponer limitaciones al reprocesado y el uso de productos reprocesados
Vemos que tenemos que esperar al desarrollo del artículo 17 de este Reglamento.
Seguimos con nuevas normas. Tras la lectura del nuevo Reglamento sobre Productos Sanitarios, parece que las centrales de esterilización van a tener un nuevo concepto ¿Cambio de paradigma? Nos convertimos en RUMED«Reprocessing Unit for Medical Devices» (Recomendaciones).
Pues resulta que tenemos que hacer «Gestión de Riesgos en la Central/RUMED», para ello nos puede ayuda la UNE-EN-ISO 13485:2016AC. Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios. Pero que nadie crea que le va a dar un protocolo o guía de actuación, sino una serie de recomendaciones para aplicarlo. Esta actualización de 2017 tiene un coste de 0€, pero que nadie se crea que AENOR regala las normas, la anterior tiene un precio de 90€ (¡a rascarse el bolsillo!).
Como suele ocurrir con las ISO, no nos dan soluciones sino un camino para empezar a trabajar.
Cuando una Central decide obtener la Licencia de Funcionamiento de acuerdo al RD 1519/2009, puede pasar a un paso más allá (de carácter voluntario) y certificarse en la UNE-EN ISO 9001:2015. Quizás lo siguiente, es que la certificación según la UNE-EN ISO 13485 nos sea obligatorio si nos queremos convertir en fabricantes.























 Es un tema tratado en este Blog varias veces (
Es un tema tratado en este Blog varias veces (




Debe estar conectado para enviar un comentario.