


Esta duda se planteó en el II Curso de Reprocesado de Instrumental Médico realizado en Oviedo del 6 al 8 de noviembre de 2005. Gracias a Jorge y Mamen por la excelente organización. Este curso tiene mucho futuro.

Sé que me estoy metiendo en un buen «freago», pero es que es un campo en continuo desarrollo y tenemos que ir aclarando conceptos. Además, esta es la entrada 300 del Blog y hay que meterse en temas controvertidos.
El RD 192/2023 regula los productos sanitarios de uso humano en España, incluyendo sus accesorios, y hará de acompañamiento al Reglamento (UE) 2017/745 (“MDR”) que es de aplicación directa en la UE.
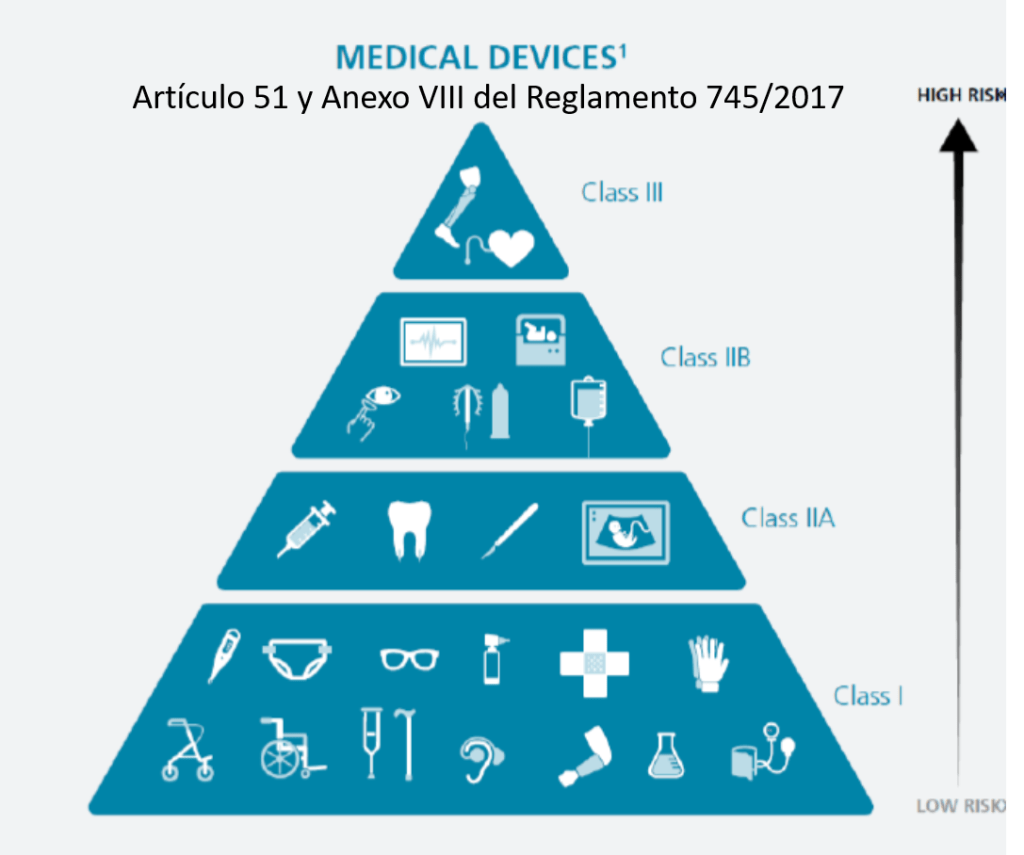
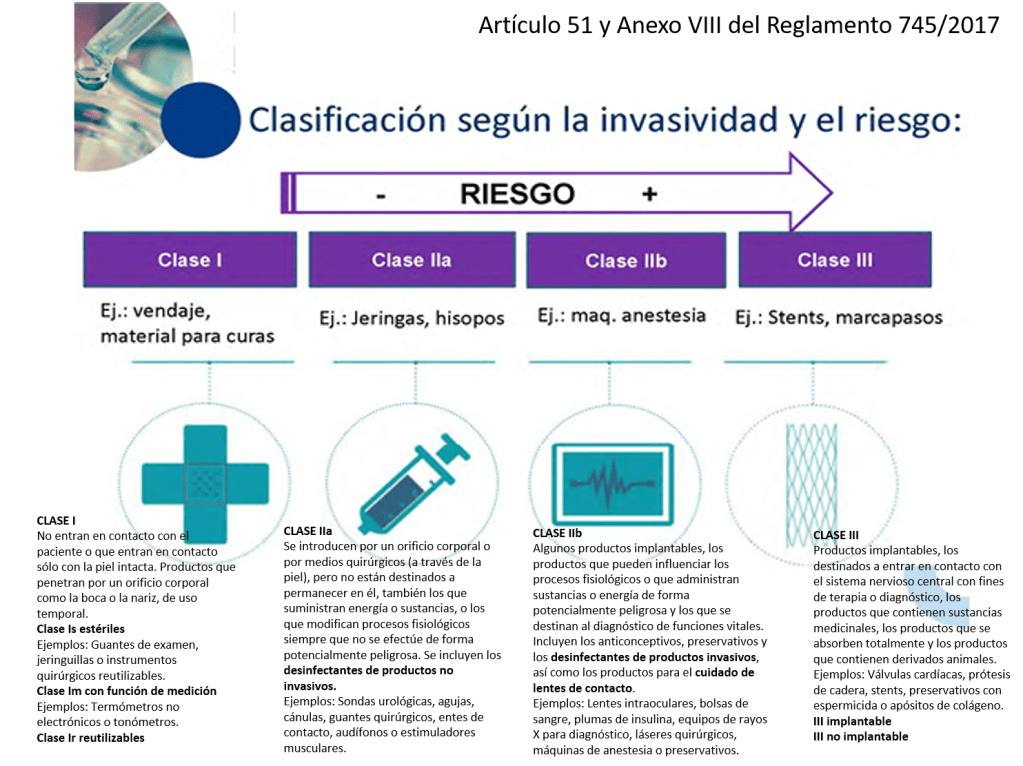
Si un hospital recibe un implante de tipo IIb o III (alto riesgo, como prótesis cardíacas, implantes neurológicos, implantes óseos, dispositivos vasculares, entre otros), y el fabricante proporciona en las IFU (Instrucciones de Uso o para el usuario final) procedimientos específicos de limpieza, reprocesamiento y esterilización, es fundamental cumplir con una serie de consideraciones técnicas y regulatorias obligatorias:
1. Validación del proceso por parte del fabricante
El proceso descrito en la IFU debe haber sido validado por el fabricante. Esto implica que el fabricante ha demostrado, mediante estudios documentados, que el reprocesamiento no compromete la seguridad, la funcionalidad ni la biocompatibilidad del dispositivo. El hospital solo podrá realizar la limpieza o esterilización conforme a esas instrucciones validadas.
Su uso está previsto una sola vez (single use), salvo indicación contraria explícita y validada por el fabricante. Y que quiere decir ésto, pues que si se te infecta o se complica la cosa, no vale con quitarlo y que nosotros lo reprocesemos otra vez (OJO CON ÉSTO).
Y aquí pregunto: ¿tenemos validadas nuestras lavadoras termodesinfectadoras? ¿y nuestros autoclaves y esterilizadores? Pues, en general, NO. Entonces, ¿podemos asegurar al usuario final que el producto final que le entregamos va a ser como él lo espera? Si en al menos uno de los dos casos tu respuesta ha sido NO, entonces no te metas en este lío. Otro OJO, validar no es poner indicadores biológicos. Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.
2. Cumplimiento estricto de las instrucciones
El establecimiento sanitario debe seguir de manera exacta todos los pasos indicados por el fabricante: tipo de detergente, método y parámetros de esterilización (temperatura, presión, tiempo, tipo de ciclo), tipo de envase o empaque, controles de esterilidad, etc. Cualquier desviación invalida la garantía del proceso y traslada la responsabilidad al hospital.
- El fabricante ha validado el proceso de reprocesamiento, incluyendo limpieza, desinfección y esterilización, y lo describe en la IFU.
- Tú (el hospital) sigues exactamente los pasos, parámetros y materiales especificados (p. ej., tipo de detergente, temperatura, método de esterilización).
- El fabricante asume la responsabilidad de que el dispositivo sigue siendo seguro y eficaz después del proceso, siempre que tú sigas sus instrucciones al pie de la letra.
👉 En este caso, el hospital no se convierte en “fabricante” ni asume responsabilidad adicional, ya que está actuando conforme al uso previsto y validado por el fabricante.

3. Responsabilidad del fabricante y del hospital
Mientras el hospital actúe conforme a las IFU, la responsabilidad de la seguridad del dispositivo tras el reprocesamiento recae en el fabricante. En cambio, si el hospital modifica el proceso, utiliza métodos no validados o reprocesa un dispositivo declarado de “un solo uso”, entonces asume el rol de fabricante según la legislación aplicable (p. ej., Reglamento (UE) 2017/745, artículo 17), con todas las obligaciones regulatorias asociadas.
Para productos implantables, el RD regula que junto al producto se debe entregar al paciente la “tarjeta de implante” que contenga información (por ejemplo: identificación del producto, número de lote, etc.). También se establecen obligaciones de registro nacional de implantes para que los centros y profesionales comuniquen datos.
4. Reprocesamiento de dispositivos de un solo uso
Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.
5. Documentación y trazabilidad
Todo procedimiento de limpieza y esterilización debe estar documentado, trazable y controlado dentro del sistema de gestión de calidad del hospital.
Esto incluye:
- Registro de lote o número de serie del dispositivo.
- Identificación del personal responsable.
- Validación y control de los equipos de reprocesamiento.
- Verificación de los parámetros del ciclo de esterilización.
| Situación | ¿Se puede limpiar/esterilizar en el hospital? | Condición |
|---|---|---|
| IFU incluye instrucciones validadas por el fabricante | ✅ Sí | Siguiendo exactamente las instrucciones validadas |
| IFU indica “single use only” o no describe reprocesamiento | ❌ No | Sería un reprocesamiento no autorizado → el hospital pasa a ser “fabricante”. |
| Implante de tipo III sin validación del proceso | ❌ No | Riesgo alto, no permitido sin validación y certificación específica. |
La entrada publicada el 06/01/2026 está teniendo cierto eco, y he recibido un comentario de Xavier Canals de Tecnomed Ingenieros Consultores que añade información que puede ser útil para todos y copio textual en cursiva:
Solo unas consideraciones regulatorias…
«1. Validación del proceso por parte del fabricante»; si no estuviera validado, no le habrían dado el marcado CE. En caso de duda sobre el proceso, solicitar información adicional al fabricante.
Un único caso donde podríamos no tener esta información es en implantes que ya vienen estériles (lo más normal) o en los implantes a medida donde el fabricante es el propio hospital, por ejemplo. (hay que solicitarselo o … no usarlo.
» Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.» Entiendo que la frase correcta es «y que puede no coincidir con la empresa fabricante del equipo»,
en general, el fabricante es el que tiene más facilidad de realizar la validación y, en su caso, reparar el equipo si no pasa la validación Debe incluir también una parte de la validación a nuestro servicio, procedimientos y registros… Imaginaros en un símil doméstico alguien que lavara prendas de color en el programa de la lavadora de algodón de 90 ºC… La lavadora, ok, pero el mal uso produce resultados defectuosos.
«4. Reprocesamiento de dispositivos de un solo uso
Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.»
El reglamento MDR indica que, al no llegar a un acuerdo de todos los países europeos, cada país debía establecer su autorización. Para España, el RD 192/2023 establece que sí se pueden reprocesar productos sanitarios de un solo uso, pero que precisan licencia, segregando el caso de «fabricante de producto reprocesado» (art.12) del de «reprocesamiento en hospitales» (art.13) y sus subcontratistas «reprocesadores externos» (art.14)
Ahora solo está permitido el de los «fabricantes de producto reprocesado», y que no tengo conocimiento de que haya ninguno autorizado. Para los hospitales, según la disposición final tercera: » 2. Las actividades de reprocesamiento de productos de un solo uso en hospitales establecidas en el capítulo III, (incluida la subcontratación de estas actividades a un reprocesador externo) requerirán el previo desarrollo por el Ministerio de Sanidad de los requisitos técnicos establecidos en este real decreto», es decir, que hay que esperar a un decreto nuevo.
y hay que tener en cuenta que el RD 192/2023: «Artículo 15. Utilización de productos de un solo uso reprocesados. … 2. No se permitirá la adquisición y utilización en España de productos que hayan sido transferidos a un tercer país para su reprocesamiento…»
Colofón final, y respuesta a la pregunta de la entrada:
¿Es posible hacerlo legalmente en España? SI
¿Es probable que tengamos todos los elementos necesarios para hacerlo? NO
Conclusión: NO, al menos de momento.
Deseo que en el día de los Reyes Magos hayáis recibido muchos regalos y no estéis leyendo esta entrada en el Blog porque os han traído la corbata o los calcetines de todos los años. Yo tengo un nuevo estuche para mi trompeta.


Espero ayudar en lo que pueda con esta entrada. Se puede liar la cosa como el que montan «El canijo de Jerez» y «Los Estanques».
Para los que me pedís algo serio:

















Debe estar conectado para enviar un comentario.