

Antes de empezar con la entrada, os recuerdo el próximo II Congreso Iberoamericano de Esterilización y el II Congreso Nacional de Reprocesamiento de Dispositivos médicos, que se desarrollará del 16 al 18 de mayo de 2024 en Cartagena de Indias (Colombia).
Aquí tenéis el enlace al Congreso y su formulario de registro al mismo.


«Me ha salido un control biológico positivo ¿Qué hago?» Esta es una pregunta que hicieron en la lista de distribución «Prevenlista» que yo coordino en España, y sé que va a traer algo de polémica y dudas como a mi me gusta, si no esto no sería divertido. Lo correcto sería decir indicador biológico, pero hay confianza para llamarlo control.
En primer lugar voy a poner el comunicado o carta que hizo SEDE, que es la Sociedad Española de Desinfección y Esterilización y que puede considerarse la voz experta en la materia en España, y que me han autorizado a publicar íntegramente en el Blog.
Nota SEDE sobre aparición de control biológico positivo
Ante la aparición de un control biológico positivo habría que tener en cuenta diferentes aspectos:
¿Realmente se dan tantas incidencias o son falsos positivos?
¿Se trata de un control por vapor, o por otro tipo de esterilización química?
¿Se usan controles biológicos rápidos?
Tenemos que exponer el siguiente contexto : la aparición de un control biológico positivo en vapor es cuanto menos anecdótico, no suele ser lo habitual. En el caso de esterilización a baja temperatura, si hay algún positivo normalmente se debe a que son de otra casa comercial distinta al químico utilizado . Esta afirmación está basada también en experiencia
Sin embargo, la mayoría de la aparición de positivos, son falsos positivos, en los que al mezclar correctamente el vial y volverlos a incubar arrojan, en efecto, resultados negativos.
Teniendo esto en cuenta, la instauración de un protocolo usando controles biológicos de lectura ultra rápida (menos de 30 minutos) no debería producir estos problemas que se mencionan, y si aun así se dieran, debería notificarse al servicio responsable del paciente en el que se ha usado el material con ese resultado, al igual que notificar a su vez al servicio de Medicina Preventiva.
No obstante consideramos conveniente, tener elaborado un protocolo de esterilización en contexto de urgencia y tener bastante material para urgencias más comunes según el alcance del hospital. Al fin y al cabo, pocos procedimientos necesitan actuación en menos de 30 minutos.
Quedamos a su disposición.
Un saludo.
Jennifer GARCIA SANCHEZ
Secretaria SEDE
Tras estos minutos musicales de buena música, que siempre me sugiere mi amigo que reside en otro continente, voy a meterme en materia.
Una de las entradas más visitadas de mi Blog es la relativa a los indicadores biológicos, donde tenéis una amplia información. Este pasado año 2023, se ha publicado bastante sobre validación y liberación paramétrica, que también voy a referenciar en esta entrada que va a ser bastante espesa.
¿Y porqué hablar de ellos? Porque la esterilización de materiales es un proceso que se denomina “Procesos especiales”, para los que no es posible la verificación de la eficacia del método en el producto final (vamos que no tenemos pruebas de que algo es estéril). La UNE 556 estableció la definición de estéril como la probabilidad de supervivencia de un microorganismo no es mayor que una entre un millón (menor que 1×106, esta expresión es lo que internacionalmente se conoce como Nivel SAL “Security Assurance Level” de 10-6).
«La esterilidad de un lote de artículos médicos es pues una noción relativa. Y según las técnicas analíticas, este es el nivel de calidad que se deberá analizar entre un millón de artículos esterilizados.» (Tomado de Manual OPS-OMS, 2008)
«Un producto se considera estéril cuando existe una probabilidad de uno entre un millón de que contenga microorganismos viables. Es lo que se llama S.A.L. (Sterility Assurance Level o Nivel Seguro de Esterilidad) y se expresa como 10-6 (10 elevado a menos 6)» (Tomado de la Guía de Estándares del Ministerio de Sanidad)

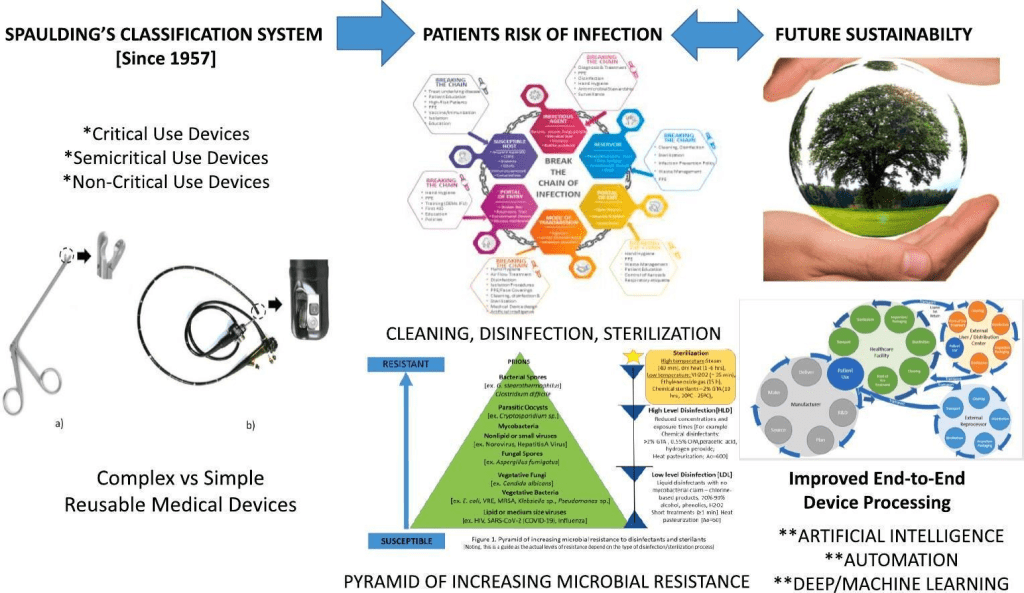
A pesar de tenerlo tan claro, aún seguimos revisando los criterios de Spaulding:
- A review of Spaulding’s classification system for effective cleaning, disinfection and sterilization of reusable medical devices: Viewed through a modern-day lens that will inform and enable future sustainability (Rowan, N. J., Kremer, T., & McDonnell, G. (2023). A review of Spaulding’s classification system for effective cleaning, disinfection and sterilization of reusable medical devices: Viewed through a modern-day lens that will inform and enable future sustainability. The Science of the total environment, 878, 162976. https://doi.org/10.1016/j.scitotenv.2023.162976). Esta revisión es excelente
- A proposed cleaning classification system for reusable medical devices to complement the Spaulding classification (Kremer, T., Rowan, N. J., & McDonnell, G. (2023). A proposed cleaning classification system for reusable medical devices to complement the Spaulding classification. The Journal of hospital infection, S0195-6701(23)00393-6. Advance online publication. https://doi.org/10.1016/j.jhin.2023.11.018)
Un indicador biológico es un dispositivo de control del proceso de esterilización que consiste de una población viable y estandarizada de microorganismos que se sabe son resistentes al proceso de esterilización. Esta forma de resistencia son esporas no patógenas llamadas Geobacillus stearothermophilus, resistentes al proceso de esterilización por vapor y Bacillus atropheus para el óxido de etileno (por ejemplo), y por lo tanto son útiles y eficaces para establecer la capacidad del ciclo de esterilización para destruir microorganismos específicos. La prueba reflejará el efecto de destrucción del proceso.
Un indicador biológico será positivo cuando exista un fallo en el proceso de esterilización. Un fallo en el proceso de esterilización incluye un mal funcionamiento del esterilizador, la calidad del vapor, si la humedad relativa del área de procesamiento no es la adecuada, el tipo y método de empaquetado, la configuración de la carga y si los parámetros del ciclo no son los apropiados para la carga que estamos esterilizando. La utilización de indicadores biológicos es una parte importante de los programas de mejora de la calidad en las organizaciones sanitarias, que garantiza que los dispositivos sanitarios estén correctamente esterilizados y debe ser implementado para la liberación del producto de la central de esterilización, en los casos que así lo requiera. La carga y los dispositivos médicos implantables deben ponerse en cuarentena hasta que estén disponibles los resultados del indicador biológico, es decir, en el caso de material implantable no hay liberación paramétrica. Si un indicador biológico es positivo, todos los dispositivos deben retirarse de las cargas procesadas desde el último indicador biológico negativo.
¿Y qué hacemos con el instrumental que hemos procesado en ese ciclo? Pues si tenemos una buena trazabilidad de instrumental, lo buscamos y retiramos.
¿Y el resto del instrumental que SI se ha utilizado? Quizás debamos echar mano de la probabilidad, o la próxima vez tener validados nuestros procesos de esterilización. ESTE ES EL PROBLEMA EN ESPAÑA, que no se exige tener validados los procesos según normas UNE-EN ISO a los equipos de las centrales de esterilización, excepto las empresas privadas que se dedican a esta labor. Hay algunos centros hospitalarios públicos y privados que han decidido validar estos procesos, pero es algo voluntario y anecdótico en el panorama español, algo impensable en Europa. Y vuelvo a insistir, las validaciones las realizan empresas externas a las nuestras y las de los fabricantes de los equipos instalados y que están acreditadas externamente. En la Guía de SEDE hay un capítulo estupendo sobre validación, y que aquí extraigo:

En este artículo de 2023 «Principles of Parametric Release: Emphasis on Data Collection and Interpretation» hace una buena revisión sobre este tema, y es muy clarificador. Viene a decir que la liberación paramétrica, que se basa en el uso de datos del proceso del producto, ofrece muchas ventajas. Sin embargo, su adopción es lenta, y la liberación suele implicar respuestas de crecimiento de indicadores biológicos y lecturas dosimétricas. Los datos proporcionados por el proceso (descritos a través de ejemplos de óxido de etileno, peróxido de hidrógeno vaporizado y radiación) pueden utilizarse mejor para informar sobre la implementación de la liberación paramétrica. Los ejemplos relativos al óxido de etileno y al peróxido de hidrógeno vaporizado demostraron la capacidad del equipo de esterilización para suministrar parámetros validados repetidamente después de que se validara la carga presentada. En los casos en los que la variabilidad de la carga no se ha tenido en cuenta en la cualificación del rendimiento, los indicadores biológicos o incluso la medición de la concentración de OE no pueden informar de forma fiable o completa sobre el impacto de dicha variabilidad en el proceso validado. El control «directo» de la concentración de OE es un requisito actual de la norma ISO 11135:2014. En consonancia con el anexo 17 de las buenas prácticas de fabricación de la Unión Europea, un requisito clave de la liberación paramétrica es disponer de datos suficientes para demostrar la repetibilidad del proceso validado.
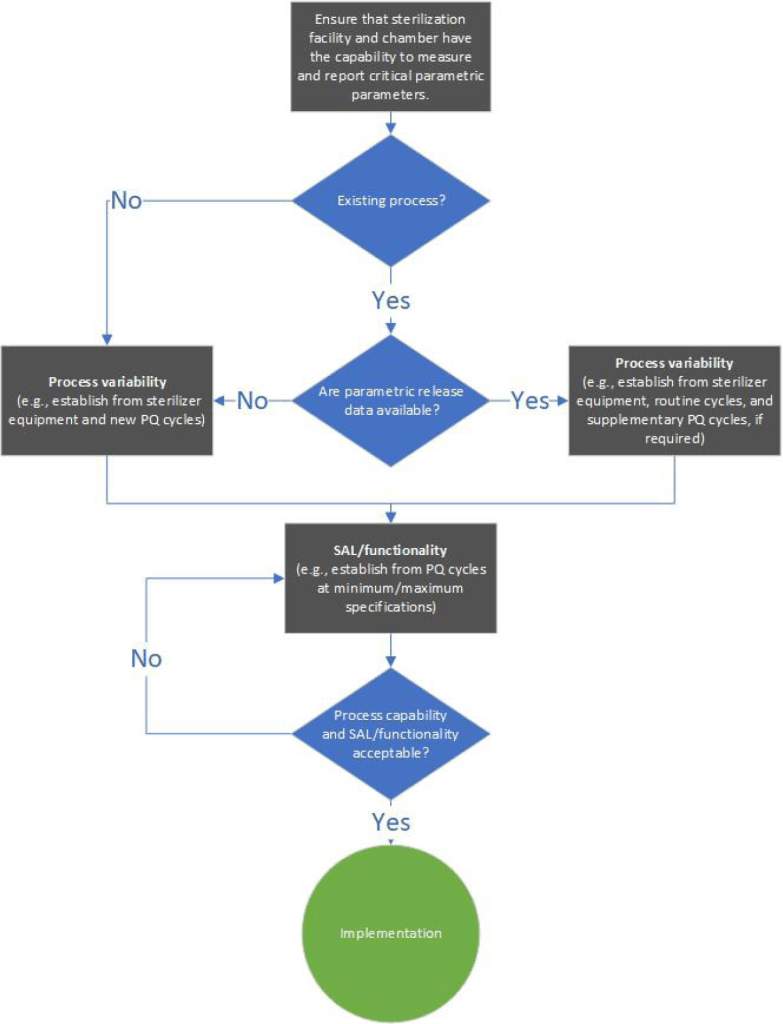
¿Qué pasa si no validamos procesos? Pues que NO deberíamos realizar una liberación paramétrica segura. La liberación paramétrica es otro término que no me canso de divulgar y decir. Si hemos validado, podemos entregar un producto sanitario sólo viendo los parámetros físicos, como son la presión, temperatura y tiempo (en el caso del vapor) y añadiendo la concentración u otra indicación (en el caso de la esterilización en frío).

La liberación paramétrica está regulada por la UNE-EN-ISO 14937:2010 que establece los requisitos generales para la caracterización de un agente esterilizante y para el desarrollo, la validación y el control de rutina de un proceso de esterilización para productos sanitarios. Los procedimientos de vapor de agua, óxido de etileno y vapor a baja temperatura con formaldehído cuentan con normas UNE específicas que regulan los requisitos específicos para su desarrollo, control y validación de su proceso de esterilización. Los procedimientos que no tengan normas específicas (vapor y plasma de peróxido de hidrógeno y ácido peracético en cámara cerrada) tienen que cumplir esta norma.

¿Nos hace falta comprar indicadores biológicos super-ultra-hiper-rápidos? Depende. Ya lo hemos hablado otras veces, depende de la rotación de instrumental, esa necesidad siempre urgente del bloque quirúrgico, o la escasez endémica de instrumental de nuestros centros. Si le preguntamos a Tamará Falcó, Marquesa de Griñón, nos dirá que quiere el instrumental quirúrgico en un nanosegundo en el metaverso (y no me lo he inventado). Si no hemos validado los autoclaves (la mayoría de los casos), nos pueden venir bien y por tanto debemos tener estos indicadores.
Quizás debamos sentarnos y pensarlo fríamente antes de decidir una compra. Si lo hemos hecho (validar equipos de esterilización) como gestores privados, fabricantes privados y algún honroso caso de la pública, nos lo podemos ahorrar. No siempre lo más rápido es lo mejor, ni lo más caro tampoco, quizás debemos mantener los orígenes.
En la Guía Suiza (de la que ya hablé) sólo mencionan los indicadores biológicos en el caso de la esterilización en frío (tomado de la entrada en el Blog):

En países como Italia y Francia no saben lo que es un Indicador Biológico. En el libro de Dominique Goullet no aparecen. Sobre los controles biológicos, dice en la página 42 que en Europa preferimos usar controles biológicos para validar un ciclo, en lugar de la liberación paramétrica (quizás no sepa que son raros los casos de validación de los equipos a vapor, al menos en España).

Y en la Guía Francesa de la SF2H de noviembre de 2022 no los mencionan, porque se validan los procesos.


Si decidimos usar indicadores biológicos debemos incubarlos en un equipo que debe tener unas condiciones técnicas óptimas de temperatura. Por cierto ¿alguna vez se comprueban esas temperaturas? No estaría mal calibrar y verificar las temperaturas de estas incubadoras mediante sondas y termómetros calibrados. Invito a todos a hacer la prueba, puede que en algún caso se lleven sorpresas, y que lo que creíamos que llegaba a 56ºC no lo alcanza, y por tanto es imposible que nos crezca o nos dé un positivo el indicador biológico. Haced la prueba, yo lo hice y la cosa no salió muy bien por cierto.
Los que no deben faltar nunca son los indicadores químicos.
¿Hay que poner un indicador biológico en cada ciclo?
Aquí hay mucha disparidad entre las Guías, en general dicen los siguiente (al menos las españolas)
- Si en el caso de la esterilización en frío.
- Si en el caso de material implantable.
- No en el caso del vapor de agua. La mayoría de las Guías recomiendan un indicador semanal (y que es una entrada del Blog actualizada), y no diario ni tampoco en cada ciclo. Evidentemente, el que te vende el indicador te dirá que lo hagas en cada ciclo, y que en cada ciclo pongas un testigo (¡ésto lo he oído de un usuario!). Que se ponga un biológico en cada ciclo, por aquello de que no tienes validado el equipo, que no tienes un mantenimiento en condiciones, y tantas cosas más que no se han hecho (y se debieran hecho), vale «aceptamos pulpo como animal de compañía», ¡pero poner un testigo en cada ciclo! Eso clama al cielo, además de suponer un derroche económico, aunque para algunos es coste efectivo. Ya sé que las empresas de indicadores biológicos me están haciendo ahora una sesión de vudú, eso pasa por no tener conflictos de intereses ni relaciones comerciales con ninguna empresa, ni monetizar este Blog.
- Por tanto, debemos repasar nuestras normas de trabajo y decidir si lo ponemos semanal como indican la mayoría de Guías o a diario (coincidiendo con un ciclo de carga), si no tenemos la confianza necesaria en que nuestro autoclave, por que no está validado o no tiene un mantenimiento preventivo bueno.
¿Qué dicen estas Guías? (y no voy a ser exhaustivo)
Os recomiendo leer las páginas 117-121 de la Guía para la normalización de los procesos en las centrales de esterilización del Servicio Gallego de Salud.


Y aquí la página 119 te dicen qué hacer si sale un biológico positivo:

Y la última parte, vuelve a insistir en la liberación paramétrica:

El apartado de Recomendaciones de esta Guía de Galicia me parece muy bueno. Además de percebes y vino rico, hacen buenas guías:

Me ha quedado una entrada un poco larga, pero creo que merece la pena perder un tiempo en intentar ayudar y clarificar.
Así que como final, pondremos algo de música freaky, extraña o no sé cómo denominarla de mis amigos «Ladilla rusa» y algo más serio.



























Debe estar conectado para enviar un comentario.