El blog de la limpieza, desinfección y esterilizacion de dispositivos sanitarios. Este Blog no pertenece ni representa a ninguna Sociedad Científica, Asociación u Organismo, su finalidad es la difusión de conocimientos y actividades relacionados con la Esterilización. Todo es fruto de una búsqueda personal de evidencia en este campo sanitario. El administrador de este blog no se responsibiliza de la información contenida en el blog pues pudieran existir errores de intepretación o traducción en algún caso de los artículos o fuentes originales. Se recomienda, por tanto, consultar con los escritos originales (enlaces), de los que tampoco este administrador se responsabiliza de su exactitud. Tampoco se responsabiliza de las opiniones vertidas por sus seguidores. Los contenidos patrocinados se indicarán debidamente.
Así titulé una ponencia en la VIII JORNADA DEL EXPERTO SOCINORTE 2022, que se celebró en el mes de mayo de 2022 en Tudela (Navarra). Ya sé que hace 1 año, pero ha habido muchas cosas interesantes entre medias.
La anterior entrada estaba dedicada al Real Decreto 192/2023 sobre reprocesado de productos de un solo uso. Tanto revuelo ha producido que la AEMPS ha sacado una Instrucción (Instrucción 1/2023) para aclararlo.
Esta vez toca hablar de la fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II).
¿Quién no ha recibido en su central alguna prótesis o producto realizado en una impresora 3D metido en una bolsa con un folio y que se lo esterilicen? No se debe hacer a la ligera si no es con unas condiciones.
Parece que podemos hacer manualidades en el hospital. Pero cuidado debe ser controlado y estudiado. Así en el trabajo de arriba se dice «Entre las desventajas del PLA destacar la baja temperatura de deformidad (55◦C) que obliga a emplear métodos de esterilización específicos, como el óxido de etileno, para evitar alterar sus propiedades si se requiere su disponibilidad intraoperatoria». Pero ya no tenemos óxido de etileno en casi ninguno de nuestros hospitales, y lo hacemos con peróxido de hidrógeno por ejemplo ¿pero son compatibles estos materiales con el peróxido, su temperatura y sus presiones? Esto nos obliga a validar los equipos, los ciclos y ver los residuos que quedan (¿cómo, consultando las UNE EN ISO correspondientes a los residuos del proceso de esterilización). Hay que decir que ese artículo es pionero (¡¡de 2016!!).
«Los modelos anatómicos, instrumental o guías que se utilizan nunca son implantados, sino que se esterilizan en el propio hospital para ser empleados en el entorno de quirófano».
Los productos sanitarios in house son aquellos productos que se fabrican y se utilizan exclusivamente en un centro sanitario. Se usan siempre y cuando no exista una alternativa de estos productos en el mercado. Un ejemplo son las prótesis hechas a partir de tecnología 3D en algunos hospitales. Se fabrican siempre bajo prescripción de un médico especialista, con características específicas y destinados a un único paciente determinado.
En el Capítulo II del Real Decreto no lo explican los artículos 9 y 10. El hospital solo podrán llevar a cabo la actividad prevista en el artículo 5.5 del Reglamento (UE) 2017/745 y cumplir todos los requisitos de control y calidad. No se aplicarán los requisitos del Reglamento, a excepción de los requisitos generales de seguridad y funcionamiento.
a) Que los productos no se cedan a otras personas jurídicas, empresas, hospitales o centros no dependientes de él. No se permitirá la venta al público de productos fabricados en hospitales. Los hospitales no podrán vender ni entregar el producto fabricado en su hospital para su uso por terceros
b) Que la fabricación y uso de los productos tengan lugar en el marco de sistemas de gestión de calidad apropiados como la UNE EN ISO 13485. Este es el proceso:
Sólo he encontrado este artículo que habla específicamente de esterilización y productos 3D. Recomiendan el óxido de etileno, y en el caso de usar gas plasma, dicen que hay que usar un relleno o no utilizarlo en el caso de dispositivos huecos. Hay más artículos sobre diferentes técnicas, materiales, medición de resultados y deformidades; pero desgraciadamente no hay ninguna norma que homogeneice estos estudios ¿debemos crear una UNE-EN ISO sobre el tema?.
c) Que el centro sanitario justifique en su documentación que no pueden satisfacerse las necesidades específicas del grupo de pacientes al que se destinan los productos, o no pueden satisfacerse con el nivel de funcionamiento adecuado, mediante otro producto equivalente comercializado.
d) Que el centro sanitario facilite a su autoridad competente información sobre el uso de dichos productos en la que se incluirá una justificación de su fabricación, modificación y uso.
f) Que el centro sanitario elabore una documentación que haga posible conocer la instalación de producción, el proceso de producción, el diseño y los datos de funcionamiento de los productos, incluida la finalidad prevista.
Además hay una serie de aspectos que conviene recordar:
Los productos de clase IIb, clase III e implantables NO podrán ser objeto de fabricación por los hospitales que desempeñen la actividad prevista en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017. Yo sigo con la duda que ya expresé en mi entrada anterior, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitará una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida pero de productos a medida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Los hospitales no podrán subcontratar ninguna de las actividades de fabricación. ¿la esterilización forma parte de la fabricación?
Se designará una persona responsable para los procedimientos que se comunicará a la Agencia Española de Medicamentos y Productos Sanitarios. Creo que hay muchos preventivistas que no saben este tema.
Parece que la pandemia nos ha hecho aprender algo. No obstante, la Agencia Española de Medicamentos y Productos Sanitarios, de forma excepcional en casos de emergencia sanitaria, podrá autorizar la fabricación de cualquier producto en centros sanitarios o institutos de salud pública en condiciones distintas a las previstas en este artículo, cuando su utilización redunde en beneficio de la salud pública o de la seguridad o salud de los pacientes.
Cada vez hay más experiencias de los hospitales, y muchos papers:
La licencia de productos a medida se basa en el Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para la concesión de licencias de funcionamiento a los fabricantes de productos sanitarios a medida. Para definir lo que es un producto sanitario a medida se va al artículo 2.a). Y este artículo 2.a) dice ««Producto sanitario», «accesorio», «producto a medida», «fabricante» y «comercialización», lo que se establece en el artículo 3 del Real Decreto 414/1996, de 1 de marzo, por el se regula los productos sanitarios». Pero es que el Real Decreto 414/1996 está derogado por el Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios; que está totalmente derogado (excepto una parte) por el Real Decreto 192/2023. Habrá que actualizar la legislación.
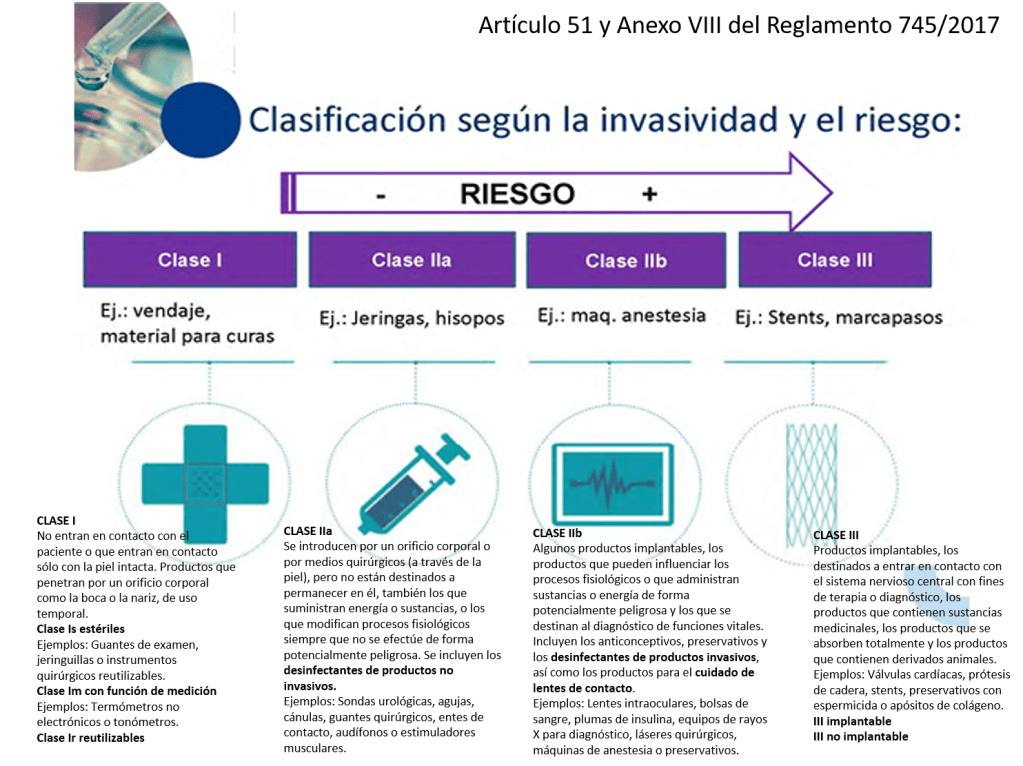
Clase I: productos que no entran en contacto con el paciente o que entran en contacto solo con la piel intacta. Productos que penetran por orificio corporal, como la boca o la nariz, de uso pasajero.
Ejemplos: productos para recolección de fluidos corporales (bolsas de orina), productos para inmovilizar partes del cuerpo o para aplicar compresión (vendas, medias elásticas), productos para el apoyo del paciente (andadores, bastones) y otros (gafas, enemas, lámparas de reconocimiento). Se excluyen de esta clase los productos que, aunque no entran en contacto con el paciente, pueden influir en procesos fisiológicos (productos que tratan la sangre destinada a reinfundirse) o los que suministran energía al cuerpo humano (equipos de radiodiagnóstico).
Dentro de la clase I podemos encontrar también:
Clase I estériles. Ejemplos: guantes de examen, jeringuillas, equipos de infusión por gravedad, gasas para proteger las heridas o para absorber exudados, instrumentos quirúrgicos reutilizables.
Clase I con función de medición. Ejemplos: jeringuillas, termómetros no electrónicos, tonómetros.
Clase IIa: se incluyen en esta clase los productos que se introducen en el cuerpo humano por orificio corporal o por medios quirúrgicos, es decir a través de la piel, pero que no están destinados a permanecer en él. También los que suministran energía o sustancias, o los que modifican procesos fisiológicos siempre y cuando no se efectúe de forma potencialmente peligrosa. También se incluyen en esta clase los desinfectantes de productos no invasivos.
Ejemplos: lentillas de contacto, los equipos de ecografía, las coronas dentales; los termómetro, circuitos de circulación extracorpórea, sondas urológicas, drenajes quirúrgicos, agujas, cánulas, guantes quirúrgicos, lentes de contacto, audífonos, estimuladores musculares: TENS, esfigmomanómetros, equipos de diagnóstico, equipos para fisioterapia.
Clase IIb: se incluyen algunos productos implantables (aunque se clasifican muchos de ellos como clase III), los productos que pueden influenciar los procesos fisiológicos o que administran sustancias o energía de forma potencialmente peligrosa y los que se destinan al diagnóstico de funciones vitales. También se clasifican como IIb los productos sanitarios anticonceptivos o para la prevención de enfermedades de transmisión sexual y los desinfectantes de productos invasivos, así como los productos para el cuidado de lentes de contacto.
Ejemplos: preservativos, los productos desinfectantes de las lentillas,lentes intraoculares, implantes de relleno tisular, suturas quirúrgicas no absorbibles, apósitos para heridas que cicatrizan por segunda intención, bolsas de sangre, hemodializadores, plumas de insulina, desfibriladores externos, equipos de rayos X para diagnóstico, láseres quirúrgicos, equipos para terapia por radiaciones, sistemas de vigilancia para cuidados intensivos, máquinas de anestesia, preservativos, etc.
Clase III: se incluyen en esta clase algunos productos implantables, los productos destinados a entrar en contacto con el sistema nervioso central o con el sistema circulatorio central con fines de terapia o diagnóstico, los productos que contienen sustancias medicinales, los productos que se absorben to-talmente y los productos que contienen derivados animales.
Ejemplos: válvulas cardíacas, prótesis de cadera, prótesis de mama, endoprótesis vasculares: stents, catéteres cardiovasculares, suturas absorbibles, adhesivos de tejidos internos biológicos, materiales de endodoncia con antibióticos, apósitos con agentes antimicrobianos.
Y muy importante recordar que los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales.
El video mató a la estrella de la radio, ¿pasará lo mismo con las impresiones 3D y el modo de operar a nuestros pacientes? ¿ha llegado la cirugía personalizada?
Aquí no acaba la entrada del Blog ¿Qué son los productos a medida? (Artículo 10 del RD 192/2023): «todo producto fabricado especialmente según la prescripción médica de cualquier persona autorizada por la legislación nacional en virtud de su cualificación profesional, en la que constan, bajo la responsabilidad de dicha persona, las características específicas de diseño, y que está destinado a ser utilizado únicamente por un paciente determinado con el fin exclusivo de atender a su estado y necesidades particulares» (artículo 2.3 del Reglamento 2017/745).
Para crear estos productos se requiere licencia previa de funcionamiento otorgada por las autoridades sanitarias de la comunidad autónoma correspondiente, de conformidad con el Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para concesión de licencias de funcionamiento para fabricantes de productos a medida. Deberán inscribirse en el Registro de responsables de la puesta en el mercado de productos a medida a través de la sede electrónica de la Agencia Española de Medicamentos y Productos Sanitarios.
Leído esto, los hospitales que impriman productos 3D específicos para un paciente deberán obtener la licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Pero claro, el Reglamento dice que no podrán ser IIb y III.
Anatomical model (CMD CLass I) for surgical planning in case of giant coronary artery bypass aneurysm manufactured with FDM technology
Surgical guide for implantology (Class IIa CMD) 3D printed in biocompatible material
Surgical guide (CMD Class IIb) in case of pectus carinatum. 3D Printed and manufactured in biocompatible material
Tomado del Webinar de la AEMPS 23/05/2023
Tomado del Webinar de la AEMPS 23/05/2023
Al final, lo que importa es ésto «The precision in planning and the perfect adaptation of both the cutting guide and the specific metal implant allowed an open approach through the upper eyelid only, so that the shortened surgical time, the minimal morbidity and the aesthetic and functional recovery of the patient have compensated the effort in the personalized design of our surgical treatment».
Realmente ha llegado la cirugía de precisión y personalizada.
La cantante Cher saben mucho de cirugía de precisión y personalizada, con sus arreglos y prótesis.
Para saber la relación de Miguel Bosé con la entrada de productos de un solo uso, deberéis llegar al final de la entrada. No haré spoiler.
Ha costado, pero ha llegado el nuevo Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. Ya está publicado en el BOE, y ¡es gratis! nadie puede decir que no lo ha leído por no estar accesible. No es magia, son tus impuestos. Nos quejamos de que las normas UNE-EN ISO son de pago, y ahora tenemos 29 folios gratuitos y accesibles, y además te avisan si se cambia algo del reglamento con el paso de tiempo si estas suscrito a «Mi BOE».
Este Real Decreto viene a esclarecer algunas cosas que no había dejado escrito el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, y del que ya hablé en este Blog.
Para no aburriros, lo que voy a hacer son pequeñas entradas de lo que me parece más importante que viene en este Real Decreto (Reglamento) que es de obligado cumplimiento en todo el territorio español (esto no entiende de autonomías) desde el 23 de marzo de 2023, día de San José Oriol. Haré entradas relativas a (este orden no es el de aparición en el Blog, si no que sigo el del reglamento):
Definiciones y aspectos a tener en cuenta (Capítulo I del Real Decreto).
Las Licencias de Funcionamiento (Capítulo II), que parece que sólo se va a pedir a las empresas que se dedican a reprocesar (empresas subcontratadas o externalizadas) o cuando un hospital sirve un producto a un hospital que no es de su grupo hospitalario (como los privados) o de su área sanitaria o un centro de centro de salud que no es de gerencia o área. Por supuesto, no puede esterilizar algo y dárselo a una clínica privada. Las licencias de funcionamiento serán necesarias en instalaciones a fabricantes, esterilizadores, agrupadores e importadores de producto sanitario y las instalaciones en que se lleven a cabo dichas actividades. Este requisito será también de aplicación a empresas que realicen un reprocesamiento de producto sanitario de un solo uso, fabricantes de productos sanitarios acogidos en el anexo XVI, aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas y la fabricación completa de los productos para terceros.
Fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II). ¿Quién no ha recibido ya alguna prótesis o producto metido en una bolsa con un folio y que se lo esterilicen? No se puede hacer si no es con unas condiciones que ya explicaré. Lo principal es que deberán realizar una comunicación previa, los `productos fabricados y utilizados en el mismo centro sanitario, los hospitales no podrán subcontratar ninguna de las actividades de fabricación, que no existan alternativas en el mercado. Y muy importante los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales. Y aquí viene algo que me llama la atención, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitar una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Reprocesado de productos de un solo uso (Capítulo III), que es de lo que hablaré hoy.
El Capítulo IV os interesa menos porque habla del Organismo notificado. Pero el Capítulo V si que interesa por que es el que trata la trazabilidad.
Y nos quedarían el VI, VII, VIII y IX que haré un resumen.
Es importante decir que las definiciones de este reglamento ya venían en el reglamento europeo. A los efectos de este real decreto, se aplicarán las definiciones recogidas en el artículo 2 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios; por lo que no hay cambios. Pero me parece importante indicar que se incluyen:
Los aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas (Art. 3.1.b). Los tatuadores y maquilladores deberán cumplir las mismas normas que la central en cuanto a seguridad. Algo hablé del tema cuando hice la entrada del brote de tiña en las peluquerías y el virus del Monkeypox.
Cuando un producto destinado por su fabricante a ser utilizado como equipo de protección individual esté destinado también a ser utilizado como producto sanitario deberá cumplir, además de la normativa aplicable a los productos de protección individual, las disposiciones de este real decreto (Art. 3.2).
La gran novedad de este reglamento es que ya se permite reprocesar productos de un solo uso. Y ya no podré cantar la canción de Camilo Sesto que tanto me gusta.
Diapositiva de una de mis ponencias
Lo he repetido mucho en este Blog que estaba prohibido reprocesa productos de un solo uso:
Diapositiva del Dr Luis Salmerón, basada en mi Blog
Y lo dice el título del Capítulo III «Reprocesamiento y nueva utilización de productos de un solo uso», habla del reprocesamiento y nueva utilización. Porque incido en lo de nueva utilización, por que mucha gente que me ha oído en foros, siempre hablo de la funcionalidad de los preservativos caducados en las carteras de los adolescentes de los años 80. Ahora tenemos que demostrar que el producto que reprocesamos funciona como si fuera nuevo. Eso no lo podemos hacer con los medios de nuestros hospitales.
En el año 2010 la Comisión Europea elaboró el Informe sobre el reprocesamiento de productos sanitarios en la Unión Europea que dice «Contrariamente a los productos sanitarios reutilizables, para los cuales se establecen requisitos en la Directiva 93/42/CEE a fin de garantizar una reutilización segura, en el caso de los productos sanitarios de un solo uso no puede afirmarse que su reutilización no presente riesgos desde el punto de vista de la salud pública. Además, también deberían tenerse en cuenta los aspectos éticos, económicos, medioambientales y relacionados con la responsabilidad del reprocesamiento de productos sanitarios de un solo uso, por lo que se analizan en mayor detalle en el presente informe».
«Los productos sanitarios de un solo uso, tales como agujas o catéteres de angioplastia, no están concebidos y diseñados para resistir a un procedimiento de reprocesamiento y el fabricante no necesita proporcionar ninguna instrucción o procedimiento validado para permitir un reprocesamiento seguro del producto, sino solamente información sobre las características o los factores técnicos conocidos por el fabricante que podrían presentar un riesgo en caso de que se reutilizara el producto. Por consiguiente, el reprocesamiento se realiza a partir de procedimientos elaborados por el usuario o el prestatario de servicios de reprocesamiento, pero sin información completa sobre el diseño y la composición del producto. Según un informe de los Países Bajos[16], la validación de un procedimiento de reprocesamiento para productos sanitarios de un solo uso, especialmente la limpieza, es una tarea que normalmente no puede realizarse en un hospital, ya que es poco probable que se disponga del equipo, el conocimiento, la experiencia y los recursos requeridos».
«No todos los productos sanitarios de un solo uso son adecuados para ser reprocesados debido a sus características o a la complejidad de determinados de estos productos. La posibilidad de reprocesamiento depende del material utilizado y de la forma del producto sanitario. Con el fin de identificar y reducir los peligros potenciales asociados con el reprocesamiento de un producto sanitario de un solo uso específico, debe evaluarse y validarse todo el ciclo de reprocesamiento que se inicia con la recogida de estos productos sanitarios de un solo uso después de su primera utilización hasta la fase final de esterilización y entrega, incluidas sus prestaciones funcionales.»
Así que el Reglamento permite reprocesa productos de un solo uso, pero NO SIGNIFICA BARRA LIBRE. Se puede hacer, pero siguiendo unas estrictas normas de calidad y seguridad «podrán llevarse a cabo siempre y cuando se cumplan los requisitos del presente real decreto» (Art. 11.1).
La gestión de riesgos: Tiene que existir un sistema. Para eso deberemos certificarnos (que es diferente a autorizarse y acreditarse) mediante alguna UNE-EN ISO como la UNE 179003 y la UNE-EN ISO 14971 (lo siento son de pago y no las puedo poner en el Blog, por que me llevarían a la cárcel). No quiero ser como Alexandra Elbakyan y su preciado sci-hub ¿Quién no lo ha utilizado alguna vez?
Un sistema de gestión de la calidad como la UNE-EN ISO 13485 (que es la adaptación al producto sanitario de la 9001).
En este tema de las UNE-EN ISO siempre puede ayudarnos (previo pago) una empresa certificadora, que generalmente les enseñas todo, les haces el trabajo y ellos dicen que haces las cosas como las describes en el manual (¡Viva los notarios del reino!). Ya sabéis lo poco que me gustan estos sistemas de auditoría por estas empresas con ánimo de lucro (incesante). Estos organismos que nos certifican deben realizarnos auditorías anuales (organismos acreditados para la certificación de sistemas de calidad de productos sanitarios) (Art. 13.5).
Tener estos dos sistema de calidad y gestión de riesgos nos obliga a validar los equipos de limpieza y termodesinfección, las selladoras y los autoclaves (frío y vapor), además de los residuos que se producen en los dispositivos médicos. TODOS. Y las validaciones cuestan mucho dinero y esfuerzo.
Trazabilidad: Eso al menos ya lo hacemos la gran mayoría de centrales o RUMED.
Notificar incidentes.
Y deberemos asegurarnos que funciona igual que antes. La funcionalidad es muy importante, y demostrarla muy difícil.
En conclusión. Los hospitales solo podrán reprocesar productos que hayan sido utilizados y reprocesados en su hospital o por un reprocesador externo incluido en su licencia. Los hospitales no podrán vender ni entregar el producto reprocesado a terceros (Art. 13.2 y 13.3).
Los hospitales podrán subcontratar las actividades de reprocesamiento a un reprocesador externo (Art. 14). OJO con estas empresas, ya que deben tener su domicilio social e instalaciones establecidas en España. Vamos que no se puede reprocesar en China o Pakistán (por ejemplo), si no que deben tener su domicilio en algún lugar del territorio español (por ejemplo en Quismondo o Pelahustán, que no son repúblicas exsocialistas soviéticas. Existen). Los reprocesadores externos deberán cumplir toda la normativa y no pueden subcontratar las actividades de reprocesamiento.
Y hay dos hechos que me parecen novedosos (Art. 15):
«Los hospitales deberán informar a los pacientes de la utilización en su hospital de productos reprocesados por su propio centro». A partir de ahora se debe informar al paciente y reflejarlo en su historia clínica.
Los productos reprocesados únicamente podrán utilizarse en los hospitales en un único paciente y durante un único proceso. Ya no vale reprocesar continuamente y a lo loco.
Una de las ventajas de permitir el reprocesado de productos de un un solo uso, sería reducir la huella de carbono de nuestros hospitales. Es un tema estudiado con los catéteres cardiológicos y catéteres de ablación, o intervenciones como las cataratas.
Cada vez serán más frecuentes los estudios de huella de carbono en hospitales o el bloque quirúrgico, endoscopias, y estudiar maneras de reducirla como la reutilización, reuso y reciclado. Quizás el medioambiente sea un motivo (y muy importante) de reprocesar productos de un solo uso .
Hay una serie de productos sanitarios que no se podrán reprocesar (Art. 11.2):
a) De clase I. Por ejemplo, bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas, guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas, tonómetros o termómetros no electrónicos.
b) A medida. OJO CON ÉSTOS
c) Fabricados y utilizados exclusivamente en hospitales de acuerdo con lo establecido en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017.
d) Que emitan radiación.
e) Utilizados para la administración de medicamentos citostáticos o radiofármacos.
f) Que incorporen sustancias medicinales.
g) Para uso en procedimientos invasivos en el sistema nervioso central.
h) Que presenten un riesgo de transmisión de encefalopatías espongiformes.
i) Implantables. OJO CON ÉSTOS, se definen como todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina aser introducido totalmente en el cuerpo humano osustituir una superficie epitelial o la superficie ocular,mediante intervención médica, y a permanecer en su lugar después de la intervención.Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
j) Relacionados con incidentes graves ocurridos tras el reprocesamiento cuya causa esté relacionada con el reprocesamiento, o para los que no pueda excluirse que la causa esté relacionada con el reprocesamiento.
k) Que tengan baterías que no puedan cambiarse o que presenten un riesgo de mal funcionamiento tras el reprocesamiento.
l) Que dispongan de un almacenamiento interno de datos necesario para el uso del producto y que no pueda cambiarse o presente un riesgo de mal funcionamiento tras el reprocesamiento.
m) Con hojas cortantes o que raspen, taladros o componentes que se desgasten que dejen de ser adecuados después del primer uso y que no puedan cambiarse o afilarse antes del siguiente procedimiento médico. OJO CON ÉSTOS, ¿COMO SABEMOS QUE LAS HOJAS NO ESTÁN AFILADAS EN UN HOSPITAL?
Así que ya sabéis, si os traen algo a reprocesar que es de un solo uso, o que se ha caducado en el almacén, o que se ha abierto por error y no se ha usado, SOLO PODÉIS REPROCESARLO SI TENÉIS LO INDICADO MÁS ARRIBA.
Y ya no es como antes, que el cirujano o traumatólogo de turno (en plan torero) te firmaba un documento, y se hacía responsable o que tu transferías la responsabilidad. Eso se ha acabado.
Para saber la relación de Miguel Bosé con la entrada de productos de un solo uso, deberéis llegar al final de la entrada. No haré spoiler.
Ha costado, pero ha llegado el nuevo Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. Ya está publicado en el BOE, y ¡es gratis! nadie puede decir que no lo ha leído por no estar accesible. No es magia, son tus impuestos. Nos quejamos de que las normas UNE-EN ISO son de pago, y ahora tenemos 29 folios gratuitos y accesibles, y además te avisan si se cambia algo del reglamento con el paso de tiempo si estas suscrito a «Mi BOE».
Este Real Decreto viene a esclarecer algunas cosas que no había dejado escrito el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, y del que ya hablé en este Blog.
Para no aburriros, lo que voy a hacer son pequeñas entradas de lo que me parece más importante que viene en este Real Decreto (Reglamento) que es de obligado cumplimiento en todo el territorio español (esto no entiende de autonomías) desde el 23 de marzo de 2023, día de San José Oriol. Haré entradas relativas a (este orden no es el de aparición en el Blog, si no que sigo el del reglamento):
Definiciones y aspectos a tener en cuenta (Capítulo I del Real Decreto).
Las Licencias de Funcionamiento (Capítulo II), que parece que sólo se va a pedir a las empresas que se dedican a reprocesar (empresas subcontratadas o externalizadas) o cuando un hospital sirve un producto a un hospital que no es de su grupo hospitalario (como los privados) o de su área sanitaria o un centro de centro de salud que no es de gerencia o área. Por supuesto, no puede esterilizar algo y dárselo a una clínica privada. Las licencias de funcionamiento serán necesarias en instalaciones a fabricantes, esterilizadores, agrupadores e importadores de producto sanitario y las instalaciones en que se lleven a cabo dichas actividades. Este requisito será también de aplicación a empresas que realicen un reprocesamiento de producto sanitario de un solo uso, fabricantes de productos sanitarios acogidos en el anexo XVI, aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas y la fabricación completa de los productos para terceros.
Fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II). ¿Quién no ha recibido ya alguna prótesis o producto metido en una bolsa con un folio y que se lo esterilicen? No se puede hacer si no es con unas condiciones que ya explicaré. Lo principal es que deberán realizar una comunicación previa, los `productos fabricados y utilizados en el mismo centro sanitario, los hospitales no podrán subcontratar ninguna de las actividades de fabricación, que no existan alternativas en el mercado. Y muy importante los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales. Y aquí viene algo que me llama la atención, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitar una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Reprocesado de productos de un solo uso (Capítulo III), que es de lo que hablaré hoy.
El Capítulo IV os interesa menos porque habla del Organismo notificado. Pero el Capítulo V si que interesa por que es el que trata la trazabilidad.
Y nos quedarían el VI, VII, VIII y IX que haré un resumen.
Es importante decir que las definiciones de este reglamento ya venían en el reglamento europeo. A los efectos de este real decreto, se aplicarán las definiciones recogidas en el artículo 2 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios; por lo que no hay cambios. Pero me parece importante indicar que se incluyen:
Los aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas (Art. 3.1.b). Los tatuadores y maquilladores deberán cumplir las mismas normas que la central en cuanto a seguridad. Algo hablé del tema cuando hice la entrada del brote de tiña en las peluquerías y el virus del Monkeypox.
Cuando un producto destinado por su fabricante a ser utilizado como equipo de protección individual esté destinado también a ser utilizado como producto sanitario deberá cumplir, además de la normativa aplicable a los productos de protección individual, las disposiciones de este real decreto (Art. 3.2).
La gran novedad de este reglamento es que ya se permite reprocesar productos de un solo uso. Y ya no podré cantar la canción de Camilo Sesto que tanto me gusta.
Diapositiva de una de mis ponencias
Lo he repetido mucho en este Blog que estaba prohibido reprocesa productos de un solo uso:
Diapositiva del Dr Luis Salmerón, basada en mi Blog
Y lo dice el título del Capítulo III «Reprocesamiento y nueva utilización de productos de un solo uso», habla del reprocesamiento y nueva utilización. Porque incido en lo de nueva utilización, por que mucha gente que me ha oído en foros, siempre hablo de la funcionalidad de los preservativos caducados en las carteras de los adolescentes de los años 80. Ahora tenemos que demostrar que el producto que reprocesamos funciona como si fuera nuevo. Eso no lo podemos hacer con los medios de nuestros hospitales.
En el año 2010 la Comisión Europea elaboró el Informe sobre el reprocesamiento de productos sanitarios en la Unión Europea que dice «Contrariamente a los productos sanitarios reutilizables, para los cuales se establecen requisitos en la Directiva 93/42/CEE a fin de garantizar una reutilización segura, en el caso de los productos sanitarios de un solo uso no puede afirmarse que su reutilización no presente riesgos desde el punto de vista de la salud pública. Además, también deberían tenerse en cuenta los aspectos éticos, económicos, medioambientales y relacionados con la responsabilidad del reprocesamiento de productos sanitarios de un solo uso, por lo que se analizan en mayor detalle en el presente informe».
«Los productos sanitarios de un solo uso, tales como agujas o catéteres de angioplastia, no están concebidos y diseñados para resistir a un procedimiento de reprocesamiento y el fabricante no necesita proporcionar ninguna instrucción o procedimiento validado para permitir un reprocesamiento seguro del producto, sino solamente información sobre las características o los factores técnicos conocidos por el fabricante que podrían presentar un riesgo en caso de que se reutilizara el producto. Por consiguiente, el reprocesamiento se realiza a partir de procedimientos elaborados por el usuario o el prestatario de servicios de reprocesamiento, pero sin información completa sobre el diseño y la composición del producto. Según un informe de los Países Bajos[16], la validación de un procedimiento de reprocesamiento para productos sanitarios de un solo uso, especialmente la limpieza, es una tarea que normalmente no puede realizarse en un hospital, ya que es poco probable que se disponga del equipo, el conocimiento, la experiencia y los recursos requeridos».
«No todos los productos sanitarios de un solo uso son adecuados para ser reprocesados debido a sus características o a la complejidad de determinados de estos productos. La posibilidad de reprocesamiento depende del material utilizado y de la forma del producto sanitario. Con el fin de identificar y reducir los peligros potenciales asociados con el reprocesamiento de un producto sanitario de un solo uso específico, debe evaluarse y validarse todo el ciclo de reprocesamiento que se inicia con la recogida de estos productos sanitarios de un solo uso después de su primera utilización hasta la fase final de esterilización y entrega, incluidas sus prestaciones funcionales.»
Así que el Reglamento permite reprocesa productos de un solo uso, pero NO SIGNIFICA BARRA LIBRE. Se puede hacer, pero siguiendo unas estrictas normas de calidad y seguridad «podrán llevarse a cabo siempre y cuando se cumplan los requisitos del presente real decreto» (Art. 11.1).
La gestión de riesgos: Tiene que existir un sistema. Para eso deberemos certificarnos (que es diferente a autorizarse y acreditarse) mediante alguna UNE-EN ISO como la UNE 179003 y la UNE-EN ISO 14971 (lo siento son de pago y no las puedo poner en el Blog, por que me llevarían a la cárcel). No quiero ser como Alexandra Elbakyan y su preciado sci-hub ¿Quién no lo ha utilizado alguna vez?
Un sistema de gestión de la calidad como la UNE-EN ISO 13485 (que es la adaptación al producto sanitario de la 9001).
En este tema de las UNE-EN ISO siempre puede ayudarnos (previo pago) una empresa certificadora, que generalmente les enseñas todo, les haces el trabajo y ellos dicen que haces las cosas como las describes en el manual (¡Viva los notarios del reino!). Ya sabéis lo poco que me gustan estos sistemas de auditoría por estas empresas con ánimo de lucro (incesante). Estos organismos que nos certifican deben realizarnos auditorías anuales (organismos acreditados para la certificación de sistemas de calidad de productos sanitarios) (Art. 13.5).
Tener estos dos sistema de calidad y gestión de riesgos nos obliga a validar los equipos de limpieza y termodesinfección, las selladoras y los autoclaves (frío y vapor), además de los residuos que se producen en los dispositivos médicos. TODOS. Y las validaciones cuestan mucho dinero y esfuerzo.
Trazabilidad: Eso al menos ya lo hacemos la gran mayoría de centrales o RUMED.
Notificar incidentes.
Y deberemos asegurarnos que funciona igual que antes. La funcionalidad es muy importante, y demostrarla muy difícil.
En conclusión. Los hospitales solo podrán reprocesar productos que hayan sido utilizados y reprocesados en su hospital o por un reprocesador externo incluido en su licencia. Los hospitales no podrán vender ni entregar el producto reprocesado a terceros (Art. 13.2 y 13.3).
Los hospitales podrán subcontratar las actividades de reprocesamiento a un reprocesador externo (Art. 14). OJO con estas empresas, ya que deben tener su domicilio social e instalaciones establecidas en España. Vamos que no se puede reprocesar en China o Pakistán (por ejemplo), si no que deben tener su domicilio en algún lugar del territorio español (por ejemplo en Quismondo o Pelahustán, que no son repúblicas exsocialistas soviéticas. Existen). Los reprocesadores externos deberán cumplir toda la normativa y no pueden subcontratar las actividades de reprocesamiento.
Y hay dos hechos que me parecen novedosos (Art. 15):
«Los hospitales deberán informar a los pacientes de la utilización en su hospital de productos reprocesados por su propio centro». A partir de ahora se debe informar al paciente y reflejarlo en su historia clínica.
Los productos reprocesados únicamente podrán utilizarse en los hospitales en un único paciente y durante un único proceso. Ya no vale reprocesar continuamente y a lo loco.
Una de las ventajas de permitir el reprocesado de productos de un un solo uso, sería reducir la huella de carbono de nuestros hospitales. Es un tema estudiado con los catéteres cardiológicos y catéteres de ablación, o intervenciones como las cataratas.
Cada vez serán más frecuentes los estudios de huella de carbono en hospitales o el bloque quirúrgico, endoscopias, y estudiar maneras de reducirla como la reutilización, reuso y reciclado. Quizás el medioambiente sea un motivo (y muy importante) de reprocesar productos de un solo uso .
Hay una serie de productos sanitarios que no se podrán reprocesar (Art. 11.2):
a) De clase I. Por ejemplo, bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas, guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas, tonómetros o termómetros no electrónicos.
b) A medida. OJO CON ÉSTOS
c) Fabricados y utilizados exclusivamente en hospitales de acuerdo con lo establecido en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017.
d) Que emitan radiación.
e) Utilizados para la administración de medicamentos citostáticos o radiofármacos.
f) Que incorporen sustancias medicinales.
g) Para uso en procedimientos invasivos en el sistema nervioso central.
h) Que presenten un riesgo de transmisión de encefalopatías espongiformes.
i) Implantables. OJO CON ÉSTOS, se definen como todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina aser introducido totalmente en el cuerpo humano osustituir una superficie epitelial o la superficie ocular,mediante intervención médica, y a permanecer en su lugar después de la intervención.Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
j) Relacionados con incidentes graves ocurridos tras el reprocesamiento cuya causa esté relacionada con el reprocesamiento, o para los que no pueda excluirse que la causa esté relacionada con el reprocesamiento.
k) Que tengan baterías que no puedan cambiarse o que presenten un riesgo de mal funcionamiento tras el reprocesamiento.
l) Que dispongan de un almacenamiento interno de datos necesario para el uso del producto y que no pueda cambiarse o presente un riesgo de mal funcionamiento tras el reprocesamiento.
m) Con hojas cortantes o que raspen, taladros o componentes que se desgasten que dejen de ser adecuados después del primer uso y que no puedan cambiarse o afilarse antes del siguiente procedimiento médico. OJO CON ÉSTOS, ¿COMO SABEMOS QUE LAS HOJAS NO ESTÁN AFILADAS EN UN HOSPITAL?
Así que ya sabéis, si os traen algo a reprocesar que es de un solo uso, o que se ha caducado en el almacén, o que se ha abierto por error y no se ha usado, SOLO PODÉIS REPROCESARLO SI TENÉIS LO INDICADO MÁS ARRIBA.
Y ya no es como antes, que el cirujano o traumatólogo de turno (en plan torero) te firmaba un documento, y se hacía responsable o que tu transferías la responsabilidad. Eso se ha acabado.
Es un día de revindicar una profesión. Ya lo hice el año pasado, y me repito éste. Quizás ya ha llegado el momento de exigir la profesionalización de los Técnicos. Hay un proyecto de Real Decreto de «Técnico en Cuidados sanitarios»; que lleva desde abril del 2019 en algún lugar del Ministerio de Educación (aunque el último borrador es de diciembre de 2019) . Pero claro, hemos tenido varios gobiernos, gobiernos en funciones y varias votaciones; y después llegó el Coronavirus. Por que la competencia es del Ministerio de Educación y Formación Profesional; mientras que el Ministerio de Sanidad tan sólo se encarga de las ordenación de las profesiones sanitarias, una vez creadas las especialidades y curriculums.
Vemos que en sus páginas 32 a 37 hay una parte importante de asepsia y antisepsia, limpieza, desinfección y esterilización. Y lo más importante, es que incide en la importancia de esta profesión fuera del hospital. Eso que llamamos los pequeños usuarios y que incluye desde centros de atención primaria (en la pública) a centros médicos privados, odontólogos, podólogos, centros de estética y tatuajes…
Hoy es un día de fiesta y reivindicación.
Pues parece que el 23 Congreso de la WFHSS será en Barcelona.
No a la guerra. Nuestro apoyo a los ciudadanos de Ucrania
«La que se avecina» es el título de la ponencia sobre el nuevo Reglamento, pero la geopolítica se ha colado y también podemos aplicarlo.
Hace unos días «tomé un café» en una sesión organizada por la casa comercial Dr. Weigert sobre el Reglamento 2017/745 (link de la noticia), del que ya hablé en el Blog en una entrada, pero esta vez lo voy a ampliar.
Lugar del encuentro
Me acompañó una amiga y profesional como Mercedes García Haro, que se centró en la aplicación directa en la RUMED. Fue de esas sesiones con amigos, en un ambiente muy distendido y agradable. Una organización inmejorable con el equipo de Dr. Weigert.
Aquí Mercedes iniciando la exposición
¿Qué debo hacer con el nuevo reglamento de productos sanitarios? (así se llamaba la charla). Leerlo y padecerlo en la intimidad como unas hemorroides.
Y aquí empezando mi charla
Este reglamento tiene como objetivo garantizar la disponibilidad en el mercado de productos sanitarios eficaces, de calidad y seguros.
De dónde venimos, y adonde vamos
Reglamento de Productos Sanitarios o Medical Devices Regulation (MDR), son dos conceptos: Reglamento y Productos sanitarios (Artículo 2 del Reglamento 745/2017).
Los reglamentos son actos jurídicos que se aplican de manera automática y uniforme en todos los países de la UE desde su entrada en vigor, sin necesidad de incorporación al derecho nacional.
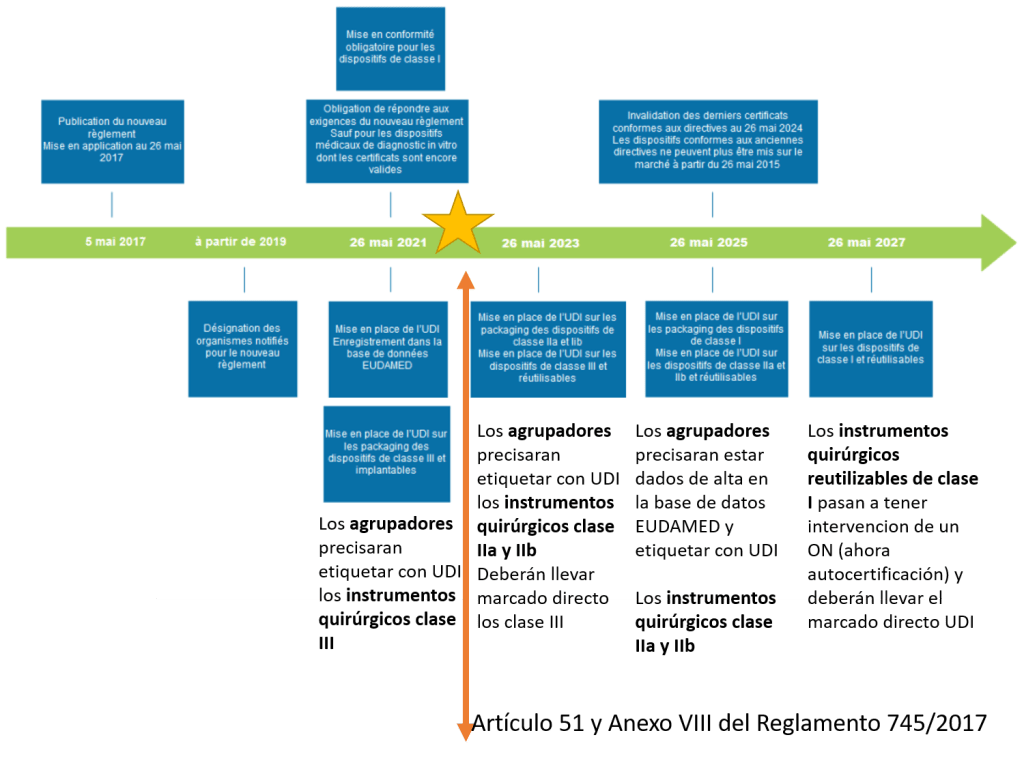
Son obligatorios, en todos sus elementos, en los Estados miembros. El Reglamento comunitario una norma de aplicación directa, el RD 1591/2009 ha pasado a quedar, en aquellos aspectos que no resulten conformes con aquel, desplazados (que no derogados). El Reglamento es una norma de aplicación directa, pero se supedita algunos aspectos o cuestiones a la regulación que se establezca a nivel nacional. El Reglamento obliga a:
1.- Derogar el Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios (excepto 21, 38, 39 y 40, y el Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos (excepto 18, 34, 35 y 36), ante la aplicación directa del Reglamento (UE) 2017/745.
2.- Desarrollar las medidas reglamentarias necesarias para aquellos aspectos en los que el reglamento comunitario ha determinado que serán los Estados miembros lo que establecerán la regulación a nivel nacional.
3.- Adaptar, adoptar o mantener las medidas requeridas por la legislación nacional.
El futuro Real Decreto es necesario para establecer:
a) Los requisitos y procedimientos para la regulación de los productos fabricados y utilizados en un centro sanitario (fabricación in house)
b) Los requisitos y procedimientos para la regulación del reprocesamiento de productos sanitarios de un solo uso
c) La regulación de la tarjeta de implantación
d) La creación de un registro nacional de comercialización de productos sanitarios
e) Establecer que la autoridad competente es la Agencia Española de Medicamentos y Productos Sanitarios independientemente de las competencias de otras autoridades sanitarias
Según el proyecto de Real Decreto: Todas las centrales de esterilización o RUMED de los hospitales (públicos y privados) tendrán que obtener la Licencia de Funcionamiento.
¿Qué es un producto sanitario?Producto sanitario (Medical Devices: MD) es cualquier instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en seres humanos y que no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.
También se considerarán productos sanitarios los productos de control o apoyo a la concepción. Y atención, los productos destinados específicamente a la limpieza, desinfección y esterilización de los productos sanitarios. Se excluyen las lentes de contacto y productos para introducirse o colocarse en los ojos, y los productos invasivos quirúrgicos para modificar anatomía o fijación partes del cuerpo, excluidos tatuajes y piercings.
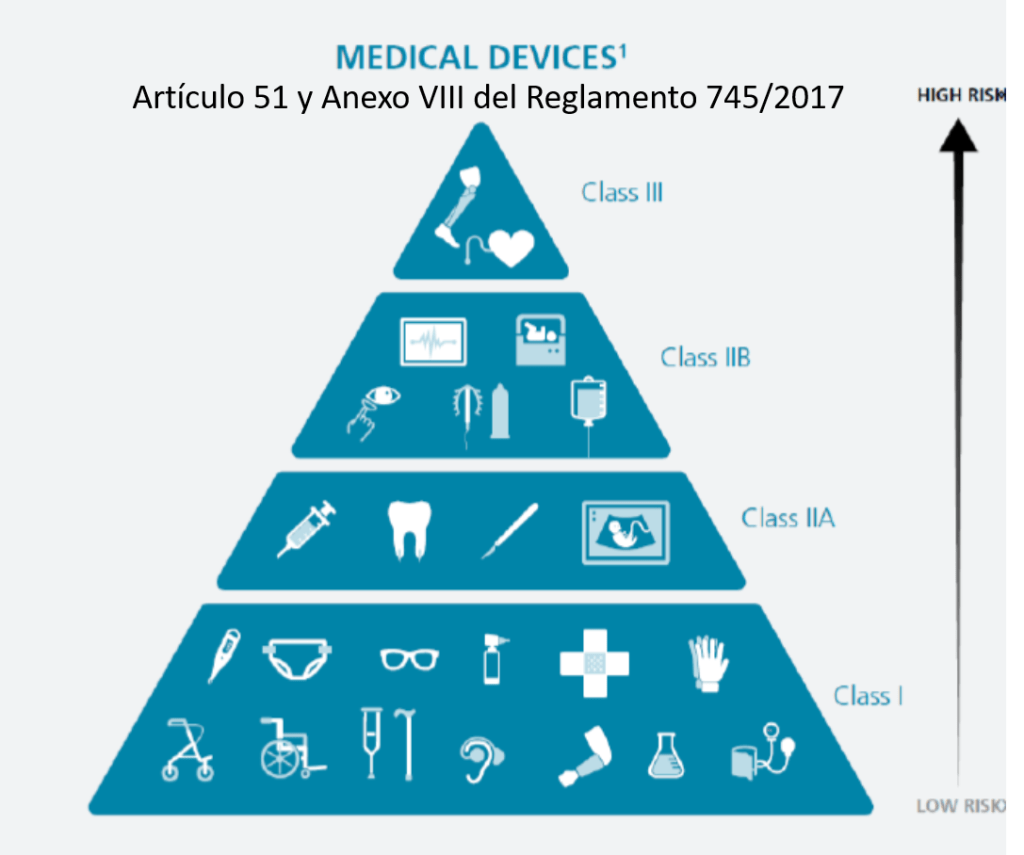
¿Cómo se clasifican los productos sanitarios? (Artículo 51 y Anexo VIII del Reglamento 745/2017)
Este reglamento tiene unos cambios relevantes que se aplican a la Central de Esterilización o RUMED:
•Los desinfectantes de productos no invasivos son IIa
•Los desinfectantes de productos invasivos son IIb
•Las lavadoras desinfectadoras pasan a ser productos sanitarios de la clase IIb
•Los equipos de esterilización pasan a ser productos sanitarios de la clase IIa
•Los indicadores químicos y biológicos NO tienen la consideración de productos sanitario (Confirmado por la AEMPS que se lo ha dicho a un amigo). La empresa GKE ha elaborado un documento donde dicen que «The GKE cleaning process monitoring indicators (CPI) are no medical devices in all countries and do not require any registration» (Párrafo actualizado 21/04/2022).
•Los productos fabricados por el propio hospital para uso interno (in house) pasan a estar regulados y precisan de documentación técnica y sistema de calidad ISO 13485 (¡¡no la 9001!!).
Aunque el Reglamento 2017/745 es de aplicación directa, hay determinados aspectos como el reprocesamiento, el régimen lingüístico o cuestiones de la fabricación en centros sanitarios, entre otros, que el reglamento determina que serán los Estados miembros los que establecerán la regulación a nivel nacional. Es por esto que en la actualidad se está elaborando el nuevo Real Decreto de Productos Sanitarios (sustituye al RD 1591/2009). Un producto sanitario:
-Se utiliza en personas.
–Finalidad prevista: Se diseña para cumplir un determinado fin. Documentada en el expediente técnico
-El fabricante lo ha validado en relación con el objetivo perseguido. Este objetivo lo establece el fabricante, y él mismo lo confirma, para emplearlo en relación con la enfermedad, discapacidad, proceso fisiológico, o patológico.
-Se ha diseñado para conseguir unos beneficios clínicos, siempre superiores a los posibles riesgos, y que él mismo ha sido capaz de evidenciar y justificar.
•Cuando se introduce un producto en el mercado, o lo pone en servicio, el fabricante se asegurará que se ha diseñado y fabricado con arreglo a los requisitos generales de producto sanitario.
•Realizar una evaluación clínica en relación con los requisitos esenciales de producto, incluyendo un seguimiento postcomercialización.
•Los de aquellos productos que no sean a medida, elaborarán y actualizarán la documentación técnica de dichos productos, permitiendo la evaluación de la conformidad de producto.
•Cumplir con las obligaciones de identificación y las de registro.
¿Cómo lo valida el fabricante? ¿Cómo el fabricante es capaz de determinar que, ciertamente, es capaz de ser eficaz y seguro para lo que él mismo pretendía Mediante el ejercicio de la evaluación clínica. La evaluación clínica es un proceso continuo, sistemático y planificado que pretende conseguir, evaluar y analizar información clínica. Estas normas pueden sernos de ayuda:
Y todo ello, en un sistema de seguridad y funcionamiento, que deberá disponer de un sistema de gestión de la calidad (UNE-EN ISO 13485 y la Guía o ayuda la UNE 179003:2009) y establecer, documentar, aplicar y mantener un sistema de gestión de riesgos (UNE-EN ISO 14971). Los estados miembros pueden permitir a los hospitales no aplicar todos los requisitos si la seguridad del nuevo producto son similares a las de los productos originales, el reprocesado se hace según especificaciones comunes sobre gestión de riesgos, la validación de procedimientos, liberación paramétrica o no de productos, ensayos, sistemas de calidad, etc… Solo se reprocesarán productos que se considere seguro hacerlo (por eso van a obligar a los hospitales obtener una Licencia de Funcionamiento). Los estados miembro pueden aplicar estas excepciones también a productos que se reprocesan en empresas externas a los hospital (PERO el producto vuelve al mismo hospital). Parece que los centros sanitarios y los fabricantes externos tendrán un régimen especial. Se abre la posibilidad a los «third party», aunque cada país puede imponer limitaciones al reprocesado y el uso de productos reprocesados. El reprocesamiento se lleva a cabo según especificaciones comunes sobre gestión de riesgos, validación de los procedimientos, liberación, sistema de gestión de la calidad, notificación de incidentes, trazabilidad.
En cuanto a los recursos humanos, se insiste en la figura del Responsable o Director Técnico, con unas funciones que ya conocemos:
•Supervisar las actividades de fabricación.
•Comprobar que se cumplen los requisitos exigidos por la reglamentación.
•Supervisar el archivo documental.
•Revisar y evaluar los incidentes.
•Ser interlocutor con las autoridades sanitarias y facilitar la documentación requerida.
•Solicitar la Licencia de Funcionamiento
Y aparece una nueva figura, que es el Responsable del cumplimiento del Reglamento (Artículo 15 Reglamento 745/2017), que tendrá esta formación, como Licenciado o Grado en Derecho, Medicina, Farmacia, Ingeniería u otra pertinente y un año experiencia en asuntos reglamentarios o SGC de productos sanitarios o cuatro de experiencia en asuntos reglamentarios o en SGC de productos sanitarios.
¿Y con todo esto ya vale?
Pues no, aun tenemos que (Artículo 10 del Reglamento 745/2017):
•Mantener a disposición de las Autoridades la documentación, la declaración de conformidad durante un periodo mínimo de 10 años desde la puesta en el mercado del último producto (15 años si son implantables).
•Disponer de un sistema de registro e información a las autoridades ante incidentes graves o acciones correctivas.
•A petición de la Autoridad competente, facilitarle toda la información para demostrar la conformidad de producto.
•En caso de que el fabricante encomiende a un tercero el diseño o fabricación del producto, se recogerá en la documentación.
•Las personas físicas o jurídicas podrán reclamar indemnizaciones por daños o perjuicios causados por un producto defectuoso; con arreglo al derecho nacional, o de la Unión, aplicable.
•Creación de una nueva base de datos llamada EUDAMED (acceso a las autoridades, la industria, los profesionales sanitarios y el público general) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
•Mejora en la trazabilidad de los productos sanitarios, mediante la implantación de un número de identificación único (UDI) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
Para terminar unas breves reflexiones en voz alta:
Todo este Reglamento puede ser un revulsivo para las RUMED, por el papel central en la seguridad del paciente que se les da, pero ¿estarán todas preparadas? Yo creo que no. Algunos hospitales o centros pequeños no tiene personal cualificado suficiente, maquinaria y equipos validados, presupuesto para desarrollarlo…
Creo que va a haber un boom de empresas especializadas en dar cobertura a todos los hospitales. Unos dando formación y asesoría en el Reglamento, sobre todo en los aspectos normativos, sistema de gestión de la calidad y sistema de gestión de riesgos.
Y otras empresas dando servicios mediante subcontratación o externalización de la RUMED o de la Dirección Técnica y Normativa.
No sabemos cómo será el futuro real decreto pero puede cambiar toda nuestra filosofía de trabajo.
Sé que es una entrada dura y con mucha información, y con muchas dudas que tendremos que ir solucionando.
El autor no tiene conflicto de intereses con la empresa Dr. Weigert
Y la despedida con uno de los mejores temas de los 70-80, con las Baccara y su «Yes Sir, I can Boogie» que se ha convertido en el himno no oficial de la selección escocesa de futbol, donde juega el petardo de Bale. En esos años, sólo sabían inglés Jesús Hermida y el Príncipe Gitano.
La asistencia sanitaria es el elemento básico de nuestra tarea como profesionales sanitarios, incluyendo la prevención, tratamiento y rehabilitación. Otros aspectos que debemos incluir en nuestro día a día son la formación continuada, la docencia y la investigación. Esto es aplicable a cualquier profesión sanitaria, que son muchas, según recoge la Ley Orgánica de las Profesiones Sanitarias (LOPS).
La calidad y seguridad del paciente son elementos básicos que debemos tener en nuestros establecimientos sanitarios, y que se unen a los principios antes enumerados. Para ello, nada mejor que realizar auditorías internas de nuestros centros, además de las inspección de apertura y periódicas que nos realizan las autoridades sanitarias.
Hoy toca hacer una auditoría de los centros de Podología o Podiatría. Para ello usaremos las Guías y Check-List del Sistema de Salud Público de Ontario, que me parecen muy claras, sencillas e intuitivas. Tenemos dos documentos (y en dos idiomas):
Check-List específico para la limpieza, desinfección y esterilización de dispositivos sanitarios; donde se evalúan los espacios, limpieza, esterilización, almacenamiento o la formación del personal (en francés).
En el Plan de Inspección de la JCCM, la esterilización aparece como uno de los puntos del mismo:
Para las personas que no conocen la profesión de Podólogo, les invito a descubrir todo el trabajo que hacen y la importancia sobre la salud general. El enlace que pongo es de la Clínica Mª del Mar Ruiz, que es una gran profesional y doctoranda mía en la Facultad de Ciencias de la Salud en Talavera de la Reina. Podéis ver las fotos del concurso que se realizó en la Facultad.
Foto del concurso de la Facultad de Ciencias de la Salud de la UCLM en Talavera de la Reina (Marta Alejandre, alumna del Grado en Podología)
Las tareas de auditoría son cada vez más frecuentes en nuestro medio, sobre todo desde el uso casi generalizado de sistemas de certificación de la calidad mediante las normas UNE-EN ISO 9001. Recuerdo siempre que certificarse es voluntario, pero lo que es obligatorio es la autorización administrativa como centro sanitario, y la autorización de la central de esterilización si servimos productos sanitarios a terceros (y dentro de poco nosotros si somos hospitales). Todavía me acuerdo, cuando me dedicaba más a temas de calidad y certificación en las centrales, que un auditor se quiso hacer «el interesante» y le tuve que recordar que yo era su cliente y quien le pagaba. La ventaja es que siempre podemos recusar a nuestros auditores de las diversas empresas de certificación que existen, no les gusta, pero no debemos olvidar que somos el cliente, el que paga, y «el cliente siempre tiene la razón».
Pronto deberemos certificarnos obligatoriamente mediante la UNE-EN ISO 13485. Esta si que me parece interesante, sobre todo si tenemos Licencia o Autorización de funcionamiento, ya que es muy fácil de obtener si hemos realizado todo el trabajo previo. Se trata de tener un sistema de gestión de la calidad sobre productos sanitarios. Ya hablaré de este tema en una entrada propia sobre el nuevo reglamento.
Otra posibilidad es cambiar de empresa certificadora buscando un mejor precio o servicio. Aquí, la cosa se convierte en un mercado persa y es muy divertido. En este mercado, cada una de las empresas te ofrece facilidades, rebajas en los precios, incluso la empresa que tienes contratada te rebaja el precio por que convierten los certificados de únicos a «multisites» y cosas que no llegas a entender. Si tienes varios centros de trabajo, al reducir el número de auditorías, se rebaja el precio. Como veis no soy un fan de las empresas de certificación y su negocio. Me gusta el aseguramiento y control de la calidad, la seguridad, la validación de equipos, la gestión de la seguridad; pero no me gustan los sistemas actuales de certificación. Al final, se certifica lo que tú pides, por ejemplo que siempre pondremos un lazo verde en un contenedor. Sabemos que no sirve de nada, pero como lo tengo puesto en mi sistema de calidad, pues me lo certifican.
Otro tema interesante de los sistemas de certificación es que tienen poca validez jurídica (en estos momentos). Si tienes un problema de responsabilidades civiles o penales, lo más seguro es que salgas más airoso del duro trance con un sistema de calidad, pero a su señoría no le va a influir mucho ese aspecto. Lo que sí va a pedir y demandar es que estemos autorizados administrativamente, y por tanto inspeccionados por la autoridad sanitaria competente (trabajo de las comunidades autónomas). En algún foro he dicho que mejor que certificarnos, sería llevar un notario a la central de esterilización y que diera fe pública de nuestros protocolos, controles, auditorías internas; con la consecuente reducción de costes y que tendría una validez jurídica pública. Cuando se hace un sorteo de un jamón o una lavadora, se suele hacer ante notario como se hacía en el programa «Un, dos, tres», no ante una empresa de certificación. Esta es mi experiencia en estos temas.
La asistencia sanitaria es el elemento básico de nuestra tarea como profesionales sanitarios, incluyendo la prevención, tratamiento y rehabilitación. Otros aspectos que debemos incluir en nuestro día a día son la formación continuada, la docencia y la investigación. Esto es aplicable a cualquier profesión sanitaria, que son muchas, según recoge la Ley Orgánica de las Profesiones Sanitarias (LOPS).
La calidad y seguridad del paciente son elementos básicos que debemos tener en nuestros establecimientos sanitarios, y que se unen a los principios antes enumerados. Para ello, nada mejor que realizar auditorías internas de nuestros centros, además de las inspección de apertura y periódicas que nos realizan las autoridades sanitarias.
Hoy toca hacer una auditoría de los centros de Podología o Podiatría. Para ello usaremos las Guías y Check-List del Sistema de Salud Público de Ontario, que me parecen muy claras, sencillas e intuitivas. Tenemos dos documentos (y en dos idiomas):
Check-List específico para la limpieza, desinfección y esterilización de dispositivos sanitarios; donde se evalúan los espacios, limpieza, esterilización, almacenamiento o la formación del personal (en francés).
Para las personas que no conocen la profesión de Podólogo, les invito a descubrir todo el trabajo que hacen y la importancia sobre la salud general. El enlace que pongo es de la Clínica Mª del Mar Ruiz, que es una gran profesional y doctoranda mía en la Facultad de Ciencias de la Salud en Talavera de la Reina. Podéis ver las fotos del concurso que se realizó en la Facultad.
Foto del concurso de la Facultad de Ciencias de la Salud de la UCLM en Talavera de la Reina ((Marta Alejandre, alumna del Grado en Podología))
Las tareas de auditoría son cada vez más frecuentes en nuestro medio, sobre todo desde el uso casi generalizado de sistemas de certificación de la calidad mediante las normas UNE-EN ISO 9001. Recuerdo siempre que certificarse es voluntario, pero lo que es obligatorio es la autorización administrativa como centro sanitario, y la autorización de la central de esterilización si servimos productos sanitarios a terceros (y dentro de poco nosotros si somos hospitales). Todavía me acuerdo, cuando me dedicaba más a temas de calidad y certificación en las centrales, que un auditor se quiso hacer «el interesante» y le tuve que recordar que yo era su cliente y quien le pagaba. La ventaja es que siempre podemos recusar a nuestros auditores de las diversas empresas de certificación que existen, no les gusta, pero no debemos olvidar que somos el cliente, el que paga, y «el cliente siempre tiene la razón».
Pronto deberemos certificarnos obligatoriamente mediante la UNE-EN ISO 13485. Esta si que me parece interesante, sobre todo si tenemos Licencia o Autorización de funcionamiento, ya que es muy fácil de obtener si hemos realizado todo el trabajo previo. Se trata de tener un sistema de gestión de la calidad sobre productos sanitarios. Ya hablaré de este tema en una entrada propia sobre el nuevo reglamento.
Otra posibilidad es cambiar de empresa certificadora buscando un mejor precio o servicio. Aquí, la cosa se convierte en un mercado persa y es muy divertido. En este mercado, cada una de las empresas te ofrece facilidades, rebajas en los precios, incluso la empresa que tienes contratada te rebaja el precio por que convierten los certificados de únicos a «multisites» y cosas que no llegas a entender. Si tienes varios centros de trabajo, al reducir el número de auditorías, se rebaja el precio. Como veis no soy un fan de las empresas de certificación y su negocio. Me gusta el aseguramiento y control de la calidad, la seguridad, la validación de equipos, la gestión de la seguridad; pero no me gustan los sistemas actuales de certificación. Al final, se certifica lo que tú pides, por ejemplo que siempre pondremos un lazo verde en un contenedor. Sabemos que no sirve de nada, pero como lo tengo puesto en mi sistema de calidad, pues me lo certifican.
Otro tema interesante de los sistemas de certificación es que tienen poca validez jurídica (en estos momentos). Si tienes un problema de responsabilidades civiles o penales, lo más seguro es que salgas más airoso del duro trance con un sistema de calidad, pero a su señoría no le va a influir mucho ese aspecto. Lo que sí va a pedir y demandar es que estemos autorizados administrativamente, y por tanto inspeccionados por la autoridad sanitaria competente (trabajo de las comunidades autónomas). En algún foro he dicho que mejor que certificarnos, sería llevar un notario a la central de esterilización y que diera fe pública de nuestros protocolos, controles, auditorías internas; con la consecuente reducción de costes y que tendría una validez jurídica pública. Cuando se hace un sorteo de un jamón o una lavadora, se suele hacer ante notario como se hacía en el programa «Un, dos, tres», no ante una empresa de certificación. Esta es mi experiencia en estos temas.
Organizadas por la Sociedad de Medicina Preventiva Hospitalaria y Asistencia Sanitaria de la Región de Murcia (SOMPRHAS), los Comités Organizador y Científico, están trabajando en el desarrollo de un Programa Científico del máximo interés, con el objetivo de actualizar conocimientos, reflexionar e intercambiar experiencias para proporcionar mayor seguridad y eficiencia en los procedimientos de limpieza, desinfección y esterilización de los dispositivos sanitarios.
Vuelvo a participar este año con este grupo de amigos y colegas de profesión en el coloquio-debate. Casi me voy a convertir en un Masterchef Celebrity.
En las Jornadas habrá dos mesas muy interesantes como es la certificación de la RUMED o UCE a través de la UNE-EN ISO 9001 y otra de riesgos laborales. En una mesa posterior se tratará el tema de auditorías internas, que son básicas para el aseguramiento de la calidad. Sobre estos sistemas de gestión tuve cierta experiencia al haber sido el Responsable de Calidad de SERMED y ESTERITEX hace ya unos años, cuando todos empezábamos con las certificaciones 9001 y 14001. De todas formas, me gustaría recalcar que lo básico y principal es obtener la autorización administrativa o Licencia de Funcionamiento a través de la AEMPS. Y la calidad está unida a la seguridad, así que se hablará de seguridad de dispositivos como los endoscopios digestivos, y la seguridad de la central y los trabajadores.
Vuestra participación es de suma importancia, por ello se os pide que colaboréis presentando trabajos en formato de Poster Digital. Este año habrá premios, se otorgarán dos premios de 150 euros uno por el comité científico y otro por votación del público patrocinados por Dr Weigert.
A través de esta web (http://www.somprhas.org/vjornadas/) podréis consultar toda la información relacionada con las jornadas, programa científico, boletín de inscripción y normativa de posters digitales. La inscripción es gratuita.
Las Jornadas van a estar acreditadas y están pendientes de la resolución para la concesión de los créditos.
También ponen a vuestra disposición el twitter de la Sociedad para que podáis seguir las últimas novedades de la asociación: @somprhas y compartir toda la información (Y el @elautoclave).
16:30 h.
Presentación e Inauguración de las Jornadas
Excmo. Sr. Juan José Pedreño Planes (por confirmar) Consejero de Salud de la Región de Murcia
Dr. Francisco Javier Campayo Rojas
Presidente SOMPRHAS.
Jefe de Sección de Medicina Preventiva.
Hospital General Universitario Reina Sofía. Murcia
Dña. Emiliana Sabuco Tébar
Vicepresidenta 2ª SOMPRHAS
16:40 h.
Mesa redonda: “Calidad y seguridad en el reprocesado de materiales sanitarios”
Moderadora:
Emiliana Sabuco Tébar
Vicepresidenta 2ª SOMPRHAS.
“Experiencia de obtención de la UNE-EN ISO 9001:2015 en la UCE”
Dña. Mireia León Castells
Enfermera Responsable de Calidad de la UCE. Hospital Universitari Germans Trias i Pujol, Badalona. GENCAT
“Programa de vigilancia y control del reprocesamiento de endoscopios digestivos”
Dra. Ángela Rincón Carlavilla
Facultativo especialista en Medicina Preventiva y Salud Pública.
Organizadas por la Sociedad de Medicina Preventiva Hospitalaria y Asistencia Sanitaria de la Región de Murcia (SOMPRHAS), los Comités Organizador y Científico, están trabajando en el desarrollo de un Programa Científico del máximo interés, con el objetivo de actualizar conocimientos, reflexionar e intercambiar experiencias para proporcionar mayor seguridad y eficiencia en los procedimientos de limpieza, desinfección y esterilización de los dispositivos sanitarios.
Vuelvo a participar este año con este grupo de amigos y colegas de profesión en el coloquio-debate. Casi me voy a convertir en un Masterchef Celebrity.
En las Jornadas habrá dos mesas muy interesantes como es la certificación de la RUMED o UCE a través de la UNE-EN ISO 9001 y otra de riesgos laborales. En una mesa posterior se tratará el tema de auditorías internas, que son básicas para el aseguramiento de la calidad. Sobre estos sistemas de gestión tuve cierta experiencia al haber sido el Responsable de Calidad de SERMED y ESTERITEX hace ya unos años, cuando todos empezábamos con las certificaciones 9001 y 14001. De todas formas, me gustaría recalcar que lo básico y principal es obtener la autorización administrativa o Licencia de Funcionamiento a través de la AEMPS. Y la calidad está unida a la seguridad, así que se hablará de seguridad de dispositivos como los endoscopios digestivos, y la seguridad de la central y los trabajadores.
Vuestra participación es de suma importancia, por ello se os pide que colaboréis presentando trabajos en formato de Poster Digital. Este año habrá premios, se otorgarán dos premios de 150 euros uno por el comité científico y otro por votación del público patrocinados por Dr Weigert.
A través de esta web (http://www.somprhas.org/vjornadas/) podréis consultar toda la información relacionada con las jornadas, programa científico, boletín de inscripción y normativa de posters digitales. La inscripción es gratuita.
Las Jornadas van a estar acreditadas y están pendientes de la resolución para la concesión de los créditos.
También ponen a vuestra disposición el twitter de la Sociedad para que podáis seguir las últimas novedades de la asociación: @somprhas y compartir toda la información (Y el @elautoclave).
16:30 h.
Presentación e Inauguración de las Jornadas
Excmo. Sr. Juan José Pedreño Planes (por confirmar) Consejero de Salud de la Región de Murcia
Dr. Francisco Javier Campayo Rojas
Presidente SOMPRHAS.
Jefe de Sección de Medicina Preventiva.
Hospital General Universitario Reina Sofía. Murcia
Dña. Emiliana Sabuco Tébar
Vicepresidenta 2ª SOMPRHAS
16:40 h.
Mesa redonda: “Calidad y seguridad en el reprocesado de materiales sanitarios”
Moderadora:
Emiliana Sabuco Tébar
Vicepresidenta 2ª SOMPRHAS.
“Experiencia de obtención de la UNE-EN ISO 9001:2015 en la UCE”
Dña. Mireia León Castells
Enfermera Responsable de Calidad de la UCE. Hospital Universitari Germans Trias i Pujol, Badalona. GENCAT
“Programa de vigilancia y control del reprocesamiento de endoscopios digestivos”
Dra. Ángela Rincón Carlavilla
Facultativo especialista en Medicina Preventiva y Salud Pública.

























































Debe estar conectado para enviar un comentario.