El blog de la limpieza, desinfección y esterilizacion de dispositivos sanitarios. Este Blog no pertenece ni representa a ninguna Sociedad Científica, Asociación u Organismo, su finalidad es la difusión de conocimientos y actividades relacionados con la Esterilización. Todo es fruto de una búsqueda personal de evidencia en este campo sanitario. El administrador de este blog no se responsibiliza de la información contenida en el blog pues pudieran existir errores de intepretación o traducción en algún caso de los artículos o fuentes originales. Se recomienda, por tanto, consultar con los escritos originales (enlaces), de los que tampoco este administrador se responsabiliza de su exactitud. Tampoco se responsabiliza de las opiniones vertidas por sus seguidores. Los contenidos patrocinados se indicarán debidamente.
Ya sabéis que soy bastante crítico con las certificaciones UNE-EN ISO o cualquier otro sistema similar. Sería largo de explicar, pero aquí va una pincelada.
Pregunta: ¿Disponer de una certificación UNE EN ISO 9001 o de la UNE EN ISO 13485 de productos sanitarios tiene alguna validez legal? ¿O te da un respaldo legal que te ayude en caso de problemas de responsabilidad en una central de esterilización de un hospital? Este asunto salió en el II Curso de Reprocesado de Instrumental Médico realizado en Oviedo del 6 al 8 de noviembre de 2005.
🔹 1. Naturaleza de las certificaciones ISO
El Real Decreto 192/2023 regula los productos sanitarios de uso humano en España, incluyendo sus accesorios, y hará de acompañamiento al Reglamento (UE) 2017/745 (“MDR”) que es de aplicación directa en la UE.
Las normas UNE-EN ISO 9001 (gestión de calidad) y UNE-EN ISO 13485 (sistemas de gestión de calidad para productos sanitarios) son normas voluntarias, no leyes. Esto significa que no tienen validez legal directa, en el sentido de que no sustituyen ni eximen del cumplimiento de la legislación sanitaria o de responsabilidad civil o penal. Y es que nos certificamos de lo que queremos, es el famoso ALCANCE, quiere decir que puedo limitar la certificación a una pequeña parte de mis procesos, y el auditor certificará que es cierto que lo hago. Puedo decir que todos mis contenedores van a salir de la central con una margarita. ¿Sirve de algo? No, pero me puedo certificar.
Sin embargo, sí tienen valor jurídico indirecto o probatorio. Vamos, que te pueden ayudar ante un juez o un problema legal.
🔹 2. Validez legal directa
Ninguna de las dos certificaciones otorga por sí misma licencia, autorización o habilitación legal para operar una central de esterilización ni para fabricar o manipular productos sanitarios. Quien lo da es el RD 192/2023.
La UNE-EN ISO 13485 puede ser requisito técnico exigido por las autoridades sanitarias o por clientes en algunos procesos de certificación de productos o contratos públicos, pero no sustituye la conformidad reglamentaria (por ejemplo, el marcado CE o las normas de higiene y trazabilidad hospitalaria).
🔹 3. Valor como respaldo legal o en el caso de problemas.
Aunque no te exime de responsabilidad, sí puede servir como elemento de defensa o atenuante en procedimientos legales o administrativos:
Demuestra diligencia y cumplimiento de estándares reconocidos internacionalmente.
Puede ayudar a probar que se aplicaron medidas razonables de control y gestión de calidad.
En un proceso judicial por una supuesta negligencia o fallo en la esterilización, tener un sistema certificado ISO 9001 o ISO 13485 puede servir como evidencia documental de que el centro tenía procedimientos controlados, auditados y validados.
👉 Es decir, no te “protege” legalmente de una demanda o sanción, pero sí mejora tu posición defensiva al mostrar diligencia debida y gestión responsable.
🔹 4. En el contexto de una central de esterilización hospitalaria
El cumplimiento de ISO 9001 o 13485 puede ayudar a garantizar trazabilidad, validación de procesos y control documental, que son claves para la seguridad del paciente.
Además, puede facilitar auditorías sanitarias o acreditaciones hospitalarias (como Joint Commission, ACSA, etc.).
Pero, ante un incidente, la responsabilidad recaería igualmente en los profesionales o la institución si se demuestra una mala praxis o incumplimiento de la normativa sanitaria.
🔹 5. Resumen
Aspecto
ISO 9001 / ISO 13485
Valor legal
Obligatoriedad
Voluntaria
No sustituye requisitos legales.
Licencia o autorización
No
Ninguna.
Valor probatorio
Sí
Sirve como evidencia de buena práctica.
Protección ante sanciones o demandas
Parcial / indirecta
Puede mitigar o apoyar la defensa.
Relevancia práctica
Alta (mejora procesos y seguridad)
Importante en auditorías o litigios.
No he localizado ninguna sentencia que establezca de forma general y vinculante que la certificación ISO 9001 o ISO 13485 exime de responsabilidad civil o penal en el ámbito sanitario. En la práctica judicial sobre responsabilidad sanitaria (negligencias, mala praxis, responsabilidad patrimonial) los tribunales valoran un conjunto de pruebas: historia clínica, protocolos, formación del personal, mantenimiento/validación de equipos, registros de procesos y, entre ellos, la existencia de sistemas de gestión o certificaciones. Una ISO puede fortalecer la defensa (demostrar diligencia y controles), pero raramente será decisiva por sí sola. Varias resoluciones y sentencias (contencioso-administrativas) reconocen que un certificado ISO 9001 puede ser un criterio válido o un medio para acreditar solvencia técnica en concursos públicos; es doctrina consolidada en materia de contratación administrativa que no obliga a excluir otros medios alternativos de acreditación. Esto muestra valor administrativo/probatorio de las ISO, pero en un contexto distinto al de la responsabilidad sanitaria.
Según la web de unos juristas dedicados a negligencias médicas (descubrí esta página en el curso con Mamen):
Disponer de una certificación UNE-EN ISO 9001 o ISO 13485 no te da inmunidad legal, pero sí constituye una prueba sólida de que la organización aplica buenas prácticas de gestión y control de calidad reconocidas internacionalmente, lo que puede ser muy útil como respaldo legal o reputacional en caso de inspecciones, auditorías o litigios.
Se me va el santo al cielo con estos temas, como Sonora Dinamita que se le perdió la cadenita a Carmen. ¡¡Cómo me gustan esas trompetas al fondo!! Se me van los pies y los dedos, y eso que solo tocan tres notas.
Para que no digáis que pongo música freaky, más de uno y una no se esperaba alos Cure en este blog:
Esta duda se planteó en el II Curso de Reprocesado de Instrumental Médico realizado en Oviedo del 6 al 8 de noviembre de 2005. Gracias a Jorge y Mamen por la excelente organización. Este curso tiene mucho futuro.
Sé que me estoy metiendo en un buen «freago», pero es que es un campo en continuo desarrollo y tenemos que ir aclarando conceptos. Además, esta es la entrada 300 del Blog y hay que meterse en temas controvertidos.
El RD 192/2023 regula los productos sanitarios de uso humano en España, incluyendo sus accesorios, y hará de acompañamiento al Reglamento (UE) 2017/745 (“MDR”) que es de aplicación directa en la UE.
Si un hospital recibe un implante de tipo IIb o III (alto riesgo, como prótesis cardíacas, implantes neurológicos, implantes óseos, dispositivos vasculares, entre otros), y el fabricante proporciona en las IFU (Instrucciones de Uso o para el usuario final)procedimientos específicos de limpieza, reprocesamiento y esterilización, es fundamental cumplir con una serie de consideraciones técnicas y regulatorias obligatorias:
1. Validación del proceso por parte del fabricante
El proceso descrito en la IFU debe haber sido validado por el fabricante. Esto implica que el fabricante ha demostrado, mediante estudios documentados, que el reprocesamiento no compromete la seguridad, la funcionalidad ni la biocompatibilidad del dispositivo. El hospital solo podrá realizar la limpieza o esterilización conforme a esas instrucciones validadas.
Su uso está previsto una sola vez (single use), salvo indicación contraria explícita y validada por el fabricante. Y que quiere decir ésto, pues que si se te infecta o se complica la cosa, no vale con quitarlo y que nosotros lo reprocesemos otra vez (OJO CON ÉSTO).
Y aquí pregunto: ¿tenemos validadas nuestras lavadoras termodesinfectadoras? ¿y nuestros autoclaves y esterilizadores? Pues, en general, NO. Entonces, ¿podemos asegurar al usuario final que el producto final que le entregamos va a ser como él lo espera? Si en al menos uno de los dos casos tu respuesta ha sido NO, entonces no te metas en este lío. Otro OJO, validar no es poner indicadores biológicos. Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.
2. Cumplimiento estricto de las instrucciones
El establecimiento sanitario debe seguir de manera exacta todos los pasos indicados por el fabricante: tipo de detergente, método y parámetros de esterilización (temperatura, presión, tiempo, tipo de ciclo), tipo de envase o empaque, controles de esterilidad, etc. Cualquier desviación invalida la garantía del proceso y traslada la responsabilidad al hospital.
El fabricante ha validado el proceso de reprocesamiento, incluyendo limpieza, desinfección y esterilización, y lo describe en la IFU.
Tú (el hospital) sigues exactamente los pasos, parámetros y materiales especificados (p. ej., tipo de detergente, temperatura, método de esterilización).
El fabricante asume la responsabilidad de que el dispositivo sigue siendo seguro y eficaz después del proceso, siempre que tú sigas sus instrucciones al pie de la letra.
👉 En este caso, el hospital no se convierte en “fabricante” ni asume responsabilidad adicional, ya que está actuando conforme al uso previsto y validado por el fabricante.
3. Responsabilidad del fabricante y del hospital
Mientras el hospital actúe conforme a las IFU, la responsabilidad de la seguridad del dispositivo tras el reprocesamiento recae en el fabricante. En cambio, si el hospital modifica el proceso, utiliza métodos no validados o reprocesa un dispositivo declarado de “un solo uso”, entonces asume el rol de fabricante según la legislación aplicable (p. ej., Reglamento (UE) 2017/745, artículo 17), con todas las obligaciones regulatorias asociadas.
Para productos implantables, el RD regula que junto al producto se debe entregar al paciente la “tarjeta de implante” que contenga información (por ejemplo: identificación del producto, número de lote, etc.). También se establecen obligaciones de registro nacional de implantes para que los centros y profesionales comuniquen datos.
4. Reprocesamiento de dispositivos de un solo uso
Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.
Todo procedimiento de limpieza y esterilización debe estar documentado, trazable y controlado dentro del sistema de gestión de calidad del hospital. Esto incluye:
Registro de lote o número de serie del dispositivo.
Identificación del personal responsable.
Validación y control de los equipos de reprocesamiento.
Verificación de los parámetros del ciclo de esterilización.
Situación
¿Se puede limpiar/esterilizar en el hospital?
Condición
IFU incluye instrucciones validadas por el fabricante
✅ Sí
Siguiendo exactamente las instrucciones validadas
IFU indica “single use only” o no describe reprocesamiento
❌ No
Sería un reprocesamiento no autorizado → el hospital pasa a ser “fabricante”.
Implante de tipo III sin validación del proceso
❌ No
Riesgo alto, no permitido sin validación y certificación específica.
La entrada publicada el 06/01/2026 está teniendo cierto eco, y he recibido un comentario de Xavier Canals de Tecnomed Ingenieros Consultores que añade información que puede ser útil para todos y copio textual en cursiva:
Solo unas consideraciones regulatorias…
«1. Validación del proceso por parte del fabricante»; si no estuviera validado, no le habrían dado el marcado CE. En caso de duda sobre el proceso, solicitar información adicional al fabricante. Un único caso donde podríamos no tener esta información es en implantes que ya vienen estériles (lo más normal) o en los implantes a medida donde el fabricante es el propio hospital, por ejemplo. (hay que solicitarselo o … no usarlo.
» Validar es certificar que un autoclave o un esterilizador cumple unas normas de validación establecidas previamente; y que debe realizar en la instalación una empresa que no puede coincidir con la empresa fabricante del equipo.» Entiendo que la frase correcta es «y que puede no coincidir con la empresa fabricante del equipo»,
en general, el fabricante es el que tiene más facilidad de realizar la validación y, en su caso, reparar el equipo si no pasa la validación Debe incluir también una parte de la validación a nuestro servicio, procedimientos y registros… Imaginaros en un símil doméstico alguien que lavara prendas de color en el programa de la lavadora de algodón de 90 ºC… La lavadora, ok, pero el mal uso produce resultados defectuosos.
«4. Reprocesamiento de dispositivos de un solo uso Los dispositivos marcados como “single use” no deben ser reprocesados ni reesterilizados. En el caso de implantes de clase IIb o III, el reprocesamiento está prohibido realizarlo (de momento), salvo que exista una autorización expresa y un proceso validado conforme a la normativa nacional o europea vigente.» El reglamento MDR indica que, al no llegar a un acuerdo de todos los países europeos, cada país debía establecer su autorización. Para España, el RD 192/2023 establece que sí se pueden reprocesar productos sanitarios de un solo uso, pero que precisan licencia, segregando el caso de «fabricante de producto reprocesado» (art.12) del de «reprocesamiento en hospitales» (art.13) y sus subcontratistas «reprocesadores externos» (art.14)
Ahora solo está permitido el de los «fabricantes de producto reprocesado», y que no tengo conocimiento de que haya ninguno autorizado. Para los hospitales, según la disposición final tercera: » 2. Las actividades de reprocesamiento de productos de un solo uso en hospitales establecidas en el capítulo III, (incluida la subcontratación de estas actividades a un reprocesador externo) requerirán el previo desarrollo por el Ministerio de Sanidad de los requisitos técnicos establecidos en este real decreto», es decir, que hay que esperar a un decreto nuevo.
y hay que tener en cuenta que el RD 192/2023: «Artículo 15. Utilización de productos de un solo uso reprocesados. … 2. No se permitirá la adquisición y utilización en España de productos que hayan sido transferidos a un tercer país para su reprocesamiento…»
Colofón final, y respuesta a la pregunta de la entrada:
¿Es posible hacerlo legalmente en España? SI
¿Es probable que tengamos todos los elementos necesarios para hacerlo? NO
Conclusión: NO, al menos de momento.
Deseo que en el día de los Reyes Magos hayáis recibido muchos regalos y no estéis leyendo esta entrada en el Blog porque os han traído la corbata o los calcetines de todos los años. Yo tengo un nuevo estuche para mi trompeta.
Os espero en el próximo concierto de año nuevoQué Melchor más guapo!
Espero ayudar en lo que pueda con esta entrada. Se puede liar la cosa como el que montan «El canijo de Jerez» y «Los Estanques».
Sé que las entradas que tienen que ver con reglamentación, normativas… son muy aburridas y tediosas. Pero es que hay que conocer la legislación vigente sobre productos sanitarios. El trabajo de las centrales de esterilización va más allá de poner un Bowie o de controles.
El Reglamento 2017/745 sobre dispositivos o productos sanitarios habla de los IFUs o instrucciones de uso (dicho en español) y de su importancia en la calidad y seguridad del paciente. En la Guía de la AEMPS nos dice que la IFUs es la información facilitada por el fabricante para informar al usuario sobre la finalidad prevista de un producto, su uso correcto y las precauciones que deban tomarse (apartado 14 del artículo 2). En esta IFU deben aparecer los medios para limpiar, desinfectar y esterilizar el material, ya que ahora mismo las supervisoras de las centrales de esterilización deben estar pidiendo y casi rogando esta información a las casas comerciales.
Todos los productos deben ir acompañados de la información sobre seguridad y funcionamiento necesaria para utilizarlos de forma segura y que ayude a identificar tanto el producto como el fabricante y/o el representante autorizado, siempre teniendo en cuenta la formación y los conocimientos de los posibles usuarios. Esta información incluye la etiqueta, el embalaje del producto y los datos en las instrucciones de uso. Como excepción a los principios generales, no se requieren instrucciones de uso para los productos de Clase I si pueden utilizarse de forma adecuada sin dichas instrucciones. Lo más probable es que se presente una excepción para los productos de Clase Ir, ya que el reprocesamiento (limpieza y esterilización) sí requerirá instrucciones. Los requisitos relativos a la información que debe suministrarse con el producto se encuentran en el anexo I, apartado 23 del capítulo III y artículo 103 del Reglamento (UE) 2017/745. En el etiquetado y las instrucciones de uso, así como en los materiales promocionales del producto, el fabricante no puede (artículo 7):
Atribuir funciones y propiedades que no tenga el producto.
Crear una impresión falsa con respecto al tratamiento o diagnóstico, funciones o propiedades que el producto no tenga.
No informar al usuario o al paciente de un riesgo probable asociado con el uso del producto en línea conforme con su finalidad prevista.
Sugerir usos para el producto distintos a los que, según el MDR, formen parte de la finalidad prevista para la que se llevó a cabo el procedimiento de evaluación de la conformidad.
El marcado CE deberá figurar también en las instrucciones de uso, así como en cualquier envase de venta.
Está prohibido poner marcas que puedan inducir a error a terceros en relación con el significado del marcado CE. Se pueden colocar otras marcas adicionales en el producto, el embalaje o las instrucciones de uso, pero estas no podrán afectar negativamente a la visibilidad o la legibilidad del marcado CE.
Especialmente importante es el contenido referente a los avisos y advertencias. En realidad, es esencial que los avisos y advertencias que el fabricante considera necesarios comunicar al usuario, estén reflejados en él. Un proceso eficaz de gestión de riesgos debe aplicar las medidas consideradas necesarias. Por orden de prioridad, primero lo harán las relacionadas con el diseño del producto sanitario. Después, las que aplican cambios en los procesos relacionados con el producto sanitario. Tras ellas, está la información para la seguridad. Esta información es el propio contenido del IFU, junto con los avisos y advertencias (warnings) que el propio fabricante considere necesarios.
Las instrucciones de uso de un producto sanitario (IFU) deben contener al menos los siguientes datos, según el producto del que se esté tratando (tomado de https://productosanitario.es/):
Denominación o nombre comercial del producto.
Nombre, nombre comercial registrado o la marca registrada del fabricante y su domicilio social.
Marcado CE del producto.
Si se trata de un producto de un solo uso o es un producto reutilizable.
Finalidad prevista del producto, indicando sus indicaciones y beneficios clínicos esperados, contraindicaciones, riesgos residuales y efectos secundarios indeseables, así como las medidas de precaución apropiadas y el grupo de pacientes al que está indicado el producto.
Forma de eliminación segura, tanto del producto como de cualquier sustancia residual.
Si el producto se suministra estéril, una indicación de su estado estéril y el método de esterilización.
Características del funcionamiento del producto.
Especificaciones que necesita el usuario para utilizar el producto de forma adecuada.
Datos sobre la preparación o manipulación del producto antes de ser utilizado.
Datos de mantenimiento, limpieza, conservación y cambio de consumibles si es necesario.
Fecha de publicación de las instrucciones de uso o, si han sido revisadas, fecha de publicación e identificador de la última revisión.
Un aviso destinado al usuario o paciente de que cualquier incidente grave relacionado con el producto debe comunicarse al fabricante y a la autoridad competente del Estado miembro en el que estén establecidos el usuario y/o el paciente.
En las instrucciones de uso estará prohibida la utilización de textos, denominaciones, marcas comerciales, fotografías e imágenes u otros signos que puedan inducir a error al usuario o al paciente en cuanto a la finalidad prevista, la seguridad y el funcionamiento del producto. El fabricante debe de tener especial cuidado al redactar las instrucciones de uso para no cometer los errores de dar a entender usos del producto diferentes a su finalidad prevista, atribuir al producto funciones y/o propiedades que no posee, crear una falsa impresión sobre tratamiento o diagnóstico, funciones o propiedades que el producto no posee y no informar al usuario o al paciente sobre los posibles riesgos que conlleva la utilización del producto conforme a su finalidad prevista.
En un estudio de APIC mediante encuesta a 1.198 preventivistas o secciones de control de infecciones, mostró que el 42% de sus centros habían sido citados (por temas legales) por no respetar una IFU, el 84% de los preventivistas tuvieron que ponerse en contacto con un fabricante para aclarar la limpieza, desinfección o esterilización adecuadas de un producto, y el 8% dieron el paso adicional de ponerse en contacto con la FDA para obtener aclaraciones sobre las instrucciones de uso.
La APIC considera que se trata de una carga inaceptable que no apoya el objetivo de prevenir la transmisión de las HAI (IRAS). La APIC pide lo siguiente:
El desarrollo de herramientas para ayudar al personal sanitario en los procesos de limpieza, desinfección y esterilización de instrumentos médicos.
Informar a los fabricantes y a la FDA sobre las IFU problemáticas.
Educar a los responsables políticos y a las organizaciones sanitarias sobre las deficiencias del marco normativo actual que limitan la capacidad de los PI para proteger a los pacientes de la transmisión de las HAI a través de dispositivos médicos.
Convocar a las organizaciones interesadas a trabajar con APIC para proponer un nuevo marco regulatorio para la limpieza, desinfección y esterilización de dispositivos médicos.
Un repositorio público de instrucciones de uso para que los usuarios tengan acceso a la información adecuada sobre los productos que ya no se fabrican y/o cuando el fabricante ya no esté en activo.
En este enlace tenéis el Informe de APIC, que es bastante extenso (está en inglés).
La Asociación Española de Enfermería Quirúrgica (AEEQ) organizó el 18 Congreso Nacional de Enfermería Quirúrgica, durante los días, 23, 24 y 25 de octubre de 2024 en Sevilla. Toda la información en este enlace.
¡Qué preciosidad de calcetines!
La mesa dedicada a la esterilización es fruto del acuerdo entre la AEEQ y SEDE. En este congreso traté el tema de la fabricación in house. Es la segunda vez que participo en un congreso de esta Asociación y es un auténtico lujo encontrarse con tantos amigos y amigas.
Se trata de un tema muy actual, y están apareciendo fabricantes «in house» por todos lados. Os recomiendo que sigáis la Unidad de Planificación Avanzada y Manufactura 3D del Hospital General Universitario Gregorio Marañón de Madrid.
Me parece importante lo que dicen en esta entrada de Linkedin: «La Unidad apuesta por modelos de subrogación (bajo nuestro sistema de calidad) y #subcontratación (bajo nuestra licencia de fabricantes), creando un entorno cooperativo donde cada empresa aporta su expertise». «La filosofía de la UPAM3D se alinea con un mapa de procesos End-to-End, abarcando desde la prescripción hasta la #vigilancia en el seguimiento de nuestros pacientes. Esto nos permite priorizar la #trazabilidad y la #seguridad del paciente, actuando como garantes de cada fase del proceso».
Quien quiera seguir profundizando y viendo que esto es muy serio, puede ver esta presentación (en francés). Pronto tendremos implantes y prótesis a la carta «Hasta ahora, los implantes vienen de serie. El paciente tiene que acostumbrarse a las medidas y tamaños estandarizados. Esta es la principal razón por la que, por ejemplo, una prótesis de rodilla se desgasta prematuramente o requiere meses de rehabilitación. El cuerpo ha de adaptarse al implante. Sin embargo, avanzamos en el desarrollo de procesos y materiales que permitan que sea al revés: que cada paciente reciba el implante que se ajusta a su cuerpo, hecho “a medida”.
MESA REDONDA 2: Actualización y reprocesado de materiales sanitarios y bioseguridad ambiental
Modera: Antonio Salmerón Gracia Supervisor de RRHH de quirófanos y esterilización. HGU Morales Meseguer. Murcia
«Surg Attack» No te aburras en el quirófano con los dispositivos 3D. Dr. Juan José Criado ÁlvarezDirector Gerente Instituto Ciencias de la Salud. Consejería de Sanidad. Autor del blog El Autoclave. Profesor Asociado Facultad CC. de la Salud. UCLM
Cuando nos enfrentemos a un producto 3D no debemos de olvidar los criterios de Spaulding y la revisión que Kremer está haciendo de los mismos, y lo que dice la normativa europea en cuanto a clasificación de los dispositivos sanitarios (nuevo Reglamento Europeo de Dispositivos Médicos, MDR 2017/745).
Se presenta un futuro prometedor con la impresión 3D, pero debemos cumplir la normativa y ofrecer un producto seguro y de calidad a nuestros pacientes. Lo que nos depara el futuro nadie lo sabe, o si…
Quien quiera conocer su futuro, que se compre un espejo
Las autoridades europeas han solicitado que se comparta con todos los usuarios posibles, para saber nuestra opinión respecto a las instrucciones de uso de los dispositivos médicos en formato electrónico. Es anónima y son 2 minutos. Es interesante poder compartir la opinión de los usuarios finales.
La Asociación Española de Enfermería Quirúrgica (AEEQ) organiza el 18 Congreso Nacional de Enfermería Quirúrgica, durante los días, 23, 24 y 25 de octubre de 2024 en Sevilla.
Una vez más, las enfermeras/os quirúrgicas/os tendrán la oportunidad de poner en común conocimientos, resultados y proyectos de investigación enfermera; además de compartir experiencias profesionales que permitan contribuir a una enfermería perioperatoria más eficiente, más segura y cercana al paciente. Todo ello desde una perspectiva global y multidisciplinar, en la que se compartirán aspectos innovadores y protocolos quirúrgicos, que, aplicados en los respectivos hospitales permitirán conseguir el recorrido profesional hacia la excelencia sanitaria. El programa científico incluirá conferencias magistrales, mesas redondas y simposios, además de diferentes talleres eminentemente prácticos en los que asistentes profundizarán en las distintas especialidades quirúrgicas. Cabe destacar el elenco de ponentes que participarán, expertos en cada uno de los temas que se abordarán durante las sesiones científicas.
Habrá una mesa dedicada a la esterilización, fruto del acuerdo entre la AEEQ y SEDE. El año pasado el congreso fue en Toledo, y ahora nos veremos en Sevilla.
MESA REDONDA 2: Actualización y reprocesado de materiales sanitarios y bioseguridad ambiental
Modera: Antonio Salmerón Gracia Supervisor de RRHH de quirófanos y esterilización. HGU Morales Meseguer. Murcia
«Surg Attack» No te aburras en el quirófano con los dispositivos 3D. Dr. Juan José Criado ÁlvarezDirector Gerente Instituto Ciencias de la Salud. Consejería de Sanidad. Autor del blog El Autoclave. Profesor Asociado Facultad CC. de la Salud. UCLM
Enfermería quirúrgica en esterilización. qué, cómo y cuándo. Mª Milagros Calvo López. Enfermera referente del Servicio de Esterilización. Hospital de la Santa Creu i San Pau. Barcelona Miembro de grupo de expertos en esterilización del COIB
Espero saludaros en Sevilla, y como ha pasado un año desde Toledo a Sevilla, este bonito tema de Luz Casal.
Aquí tenéis el enlace al Congreso y su formulario de registro al mismo.
«Me ha salido un control biológico positivo ¿Qué hago?» Esta es una pregunta que hicieron en la lista de distribución «Prevenlista» que yo coordino en España, y sé que va a traer algo de polémica y dudas como a mi me gusta, si no esto no sería divertido. Lo correcto sería decir indicador biológico, pero hay confianza para llamarlo control.
En primer lugar voy a poner el comunicado o carta que hizo SEDE, que es la Sociedad Española de Desinfección y Esterilización y que puede considerarse la voz experta en la materia en España, y que me han autorizado a publicar íntegramente en el Blog.
Nota SEDE sobre aparición de control biológico positivo
Ante la aparición de un control biológico positivo habría que tener en cuenta diferentes aspectos: ¿Realmente se dan tantas incidencias o son falsos positivos? ¿Se trata de un control por vapor, o por otro tipo de esterilización química? ¿Se usan controles biológicos rápidos? Tenemos que exponer el siguiente contexto : la aparición de un control biológico positivo en vapor es cuanto menos anecdótico, no suele ser lo habitual. En el caso de esterilización a baja temperatura, si hay algún positivo normalmente se debe a que son de otra casa comercial distinta al químico utilizado . Esta afirmación está basada también en experiencia Sin embargo, la mayoría de la aparición de positivos, son falsos positivos, en los que al mezclar correctamente el vial y volverlos a incubar arrojan, en efecto, resultados negativos. Teniendo esto en cuenta, la instauración de un protocolo usando controles biológicos de lectura ultra rápida (menos de 30 minutos) no debería producir estos problemas que se mencionan, y si aun así se dieran, debería notificarse al servicio responsable del paciente en el que se ha usado el material con ese resultado, al igual que notificar a su vez al servicio de Medicina Preventiva. No obstante consideramos conveniente, tener elaborado un protocolo de esterilización en contexto de urgencia y tener bastante material para urgencias más comunes según el alcance del hospital. Al fin y al cabo, pocos procedimientos necesitan actuación en menos de 30 minutos. Quedamos a su disposición. Un saludo. Jennifer GARCIA SANCHEZ Secretaria SEDE
Tras estos minutos musicales de buena música, que siempre me sugiere mi amigo que reside en otro continente, voy a meterme en materia.
Una de las entradas más visitadas de mi Blog es la relativa a los indicadores biológicos, donde tenéis una amplia información. Este pasado año 2023, se ha publicado bastante sobre validación y liberación paramétrica, que también voy a referenciar en esta entrada que va a ser bastante espesa.
¿Y porqué hablar de ellos? Porque la esterilización de materiales es un proceso que se denomina “Procesos especiales”, para los que no es posible la verificación de la eficacia del método en el producto final (vamos que no tenemos pruebas de que algo es estéril). La UNE 556 estableció la definición de estéril como la probabilidad de supervivencia de un microorganismo no es mayor que una entre un millón (menor que 1×106, esta expresión es lo que internacionalmente se conoce como Nivel SAL “Security Assurance Level” de 10-6).
«La esterilidad de un lote de artículos médicos es pues una noción relativa. Y según las técnicas analíticas, este es el nivel de calidad que se deberá analizar entre un millón de artículos esterilizados.» (Tomado de Manual OPS-OMS, 2008)
«Un producto se considera estéril cuando existe una probabilidad de uno entre un millón de que contenga microorganismos viables. Es lo que se llama S.A.L. (Sterility Assurance Level o Nivel Seguro de Esterilidad) y se expresa como 10-6 (10 elevado a menos 6)» (Tomado de la Guía de Estándares del Ministerio de Sanidad)
Un indicador biológico es un dispositivo de control del proceso de esterilización que consiste de una población viable y estandarizada de microorganismos que se sabe son resistentes al proceso de esterilización. Esta forma de resistencia son esporas no patógenas llamadas Geobacillus stearothermophilus, resistentes al proceso de esterilización por vapor y Bacillus atropheus para el óxido de etileno (por ejemplo), y por lo tanto son útiles y eficaces para establecer la capacidad del ciclo de esterilización para destruir microorganismos específicos. La prueba reflejará el efecto de destrucción del proceso.
Un indicador biológico será positivo cuando exista un fallo en el proceso de esterilización. Un fallo en el proceso de esterilización incluye un mal funcionamiento del esterilizador, la calidad del vapor, si la humedad relativa del área de procesamiento no es la adecuada, el tipo y método de empaquetado, la configuración de la carga y si los parámetros del ciclo no son los apropiados para la carga que estamos esterilizando. La utilización de indicadores biológicos es una parte importante de los programas de mejora de la calidad en las organizaciones sanitarias, que garantiza que los dispositivos sanitarios estén correctamente esterilizados y debe ser implementado para la liberación del producto de la central de esterilización, en los casos que así lo requiera. La carga y los dispositivos médicos implantables deben ponerse en cuarentena hasta que estén disponibles los resultados del indicador biológico, es decir, en el caso de material implantable no hay liberación paramétrica. Si un indicador biológico es positivo, todos los dispositivos deben retirarse de las cargas procesadas desde el último indicador biológico negativo.
¿Y qué hacemos con el instrumental que hemos procesado en ese ciclo? Pues si tenemos una buena trazabilidad de instrumental, lo buscamos y retiramos.
¿Y el resto del instrumental que SI se ha utilizado? Quizás debamos echar mano de la probabilidad, o la próxima vez tener validados nuestros procesos de esterilización. ESTE ES EL PROBLEMA EN ESPAÑA, que no se exige tener validados los procesos según normas UNE-EN ISO a los equipos de las centrales de esterilización, excepto las empresas privadas que se dedican a esta labor. Hay algunos centros hospitalarios públicos y privados que han decidido validar estos procesos, pero es algo voluntario y anecdótico en el panorama español, algo impensable en Europa. Y vuelvo a insistir, las validaciones las realizan empresas externas a las nuestras y las de los fabricantes de los equipos instalados y que están acreditadas externamente. En la Guía de SEDE hay un capítulo estupendo sobre validación, y que aquí extraigo:
En este artículo de 2023 «Principles of Parametric Release: Emphasis on Data Collection and Interpretation» hace una buena revisión sobre este tema, y es muy clarificador. Viene a decir que la liberación paramétrica, que se basa en el uso de datos del proceso del producto, ofrece muchas ventajas. Sin embargo, su adopción es lenta, y la liberación suele implicar respuestas de crecimiento de indicadores biológicos y lecturas dosimétricas. Los datos proporcionados por el proceso (descritos a través de ejemplos de óxido de etileno, peróxido de hidrógeno vaporizado y radiación) pueden utilizarse mejor para informar sobre la implementación de la liberación paramétrica. Los ejemplos relativos al óxido de etileno y al peróxido de hidrógeno vaporizado demostraron la capacidad del equipo de esterilización para suministrar parámetros validados repetidamente después de que se validara la carga presentada. En los casos en los que la variabilidad de la carga no se ha tenido en cuenta en la cualificación del rendimiento, los indicadores biológicos o incluso la medición de la concentración de OE no pueden informar de forma fiable o completa sobre el impacto de dicha variabilidad en el proceso validado. El control «directo» de la concentración de OE es un requisito actual de la norma ISO 11135:2014. En consonancia con el anexo 17 de las buenas prácticas de fabricación de la Unión Europea, un requisito clave de la liberación paramétrica es disponer de datos suficientes para demostrar la repetibilidad del proceso validado.
¿Qué pasa si no validamos procesos? Pues que NO deberíamos realizar una liberación paramétrica segura. La liberación paramétrica es otro término que no me canso de divulgar y decir. Si hemos validado, podemos entregar un producto sanitario sólo viendo los parámetros físicos, como son la presión, temperatura y tiempo (en el caso del vapor) y añadiendo la concentración u otra indicación (en el caso de la esterilización en frío).
La liberación paramétrica está regulada por la UNE-EN-ISO 14937:2010 que establece los requisitos generales para la caracterización de un agente esterilizante y para el desarrollo, la validación y el control de rutina de un proceso de esterilización para productos sanitarios. Los procedimientos de vapor de agua, óxido de etileno y vapor a baja temperatura con formaldehído cuentan con normas UNE específicas que regulan los requisitos específicos para su desarrollo, control y validación de su proceso de esterilización. Los procedimientos que no tengan normas específicas (vapor y plasma de peróxido de hidrógeno y ácido peracético en cámara cerrada) tienen que cumplir esta norma.
¿Nos hace falta comprar indicadores biológicos super-ultra-hiper-rápidos? Depende. Ya lo hemos hablado otras veces, depende de la rotación de instrumental, esa necesidad siempre urgente del bloque quirúrgico, o la escasez endémica de instrumental de nuestros centros. Si le preguntamos a Tamará Falcó, Marquesa de Griñón, nos dirá que quiere el instrumental quirúrgico en un nanosegundo en el metaverso (y no me lo he inventado). Si no hemos validado los autoclaves (la mayoría de los casos), nos pueden venir bien y por tanto debemos tener estos indicadores.
Quizás debamos sentarnos y pensarlo fríamente antes de decidir una compra. Si lo hemos hecho (validar equipos de esterilización) como gestores privados, fabricantes privados y algún honroso caso de la pública, nos lo podemos ahorrar. No siempre lo más rápido es lo mejor, ni lo más caro tampoco, quizás debemos mantener los orígenes.
En la Guía Suiza (de la que ya hablé) sólo mencionan los indicadores biológicos en el caso de la esterilización en frío (tomado de la entrada en el Blog):
En países como Italia y Francia no saben lo que es un Indicador Biológico. En el libro de Dominique Goullet no aparecen. Sobre los controles biológicos, dice en la página 42 que en Europa preferimos usar controles biológicos para validar un ciclo, en lugar de la liberación paramétrica (quizás no sepa que son raros los casos de validación de los equipos a vapor, al menos en España).
Si decidimos usar indicadores biológicos debemos incubarlos en un equipo que debe tener unas condiciones técnicas óptimas de temperatura. Por cierto ¿alguna vez se comprueban esas temperaturas? No estaría mal calibrar y verificar las temperaturas de estas incubadoras mediante sondas y termómetros calibrados. Invito a todos a hacer la prueba, puede que en algún caso se lleven sorpresas, y que lo que creíamos que llegaba a 56ºC no lo alcanza, y por tanto es imposible que nos crezca o nos dé un positivo el indicador biológico. Haced la prueba, yo lo hice y la cosa no salió muy bien por cierto.
¿Hay que poner un indicador biológico en cada ciclo?
Aquí hay mucha disparidad entre las Guías, en general dicen los siguiente (al menos las españolas)
Si en el caso de la esterilización en frío.
Si en el caso de material implantable.
No en el caso del vapor de agua. La mayoría de las Guías recomiendan un indicador semanal (y que es una entrada del Blog actualizada), y no diario ni tampoco en cada ciclo. Evidentemente, el que te vende el indicador te dirá que lo hagas en cada ciclo, y que en cada ciclo pongas un testigo (¡ésto lo he oído de un usuario!). Que se ponga un biológico en cada ciclo, por aquello de que no tienes validado el equipo, que no tienes un mantenimiento en condiciones, y tantas cosas más que no se han hecho (y se debieran hecho), vale «aceptamos pulpo como animal de compañía», ¡pero poner un testigo en cada ciclo! Eso clama al cielo, además de suponer un derroche económico, aunque para algunos es coste efectivo. Ya sé que las empresas de indicadores biológicos me están haciendo ahora una sesión de vudú, eso pasa por no tener conflictos de intereses ni relaciones comerciales con ninguna empresa, ni monetizar este Blog.
Por tanto, debemos repasar nuestras normas de trabajo y decidir si lo ponemos semanal como indican la mayoría de Guías o a diario (coincidiendo con un ciclo de carga), si no tenemos la confianza necesaria en que nuestro autoclave, por que no está validado o no tiene un mantenimiento preventivo bueno.
¿Qué dicen estas Guías? (y no voy a ser exhaustivo)
La anterior entrada estaba dedicada al Real Decreto 192/2023 sobre reprocesado de productos de un solo uso. Tanto revuelo ha producido que la AEMPS ha sacado una Instrucción (Instrucción 1/2023) para aclararlo.
Esta vez toca hablar de la fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II).
¿Quién no ha recibido en su central alguna prótesis o producto realizado en una impresora 3D metido en una bolsa con un folio y que se lo esterilicen? No se debe hacer a la ligera si no es con unas condiciones.
Parece que podemos hacer manualidades en el hospital. Pero cuidado debe ser controlado y estudiado. Así en el trabajo de arriba se dice «Entre las desventajas del PLA destacar la baja temperatura de deformidad (55◦C) que obliga a emplear métodos de esterilización específicos, como el óxido de etileno, para evitar alterar sus propiedades si se requiere su disponibilidad intraoperatoria». Pero ya no tenemos óxido de etileno en casi ninguno de nuestros hospitales, y lo hacemos con peróxido de hidrógeno por ejemplo ¿pero son compatibles estos materiales con el peróxido, su temperatura y sus presiones? Esto nos obliga a validar los equipos, los ciclos y ver los residuos que quedan (¿cómo, consultando las UNE EN ISO correspondientes a los residuos del proceso de esterilización). Hay que decir que ese artículo es pionero (¡¡de 2016!!).
«Los modelos anatómicos, instrumental o guías que se utilizan nunca son implantados, sino que se esterilizan en el propio hospital para ser empleados en el entorno de quirófano».
Los productos sanitarios in house son aquellos productos que se fabrican y se utilizan exclusivamente en un centro sanitario. Se usan siempre y cuando no exista una alternativa de estos productos en el mercado. Un ejemplo son las prótesis hechas a partir de tecnología 3D en algunos hospitales. Se fabrican siempre bajo prescripción de un médico especialista, con características específicas y destinados a un único paciente determinado.
En el Capítulo II del Real Decreto no lo explican los artículos 9 y 10. El hospital solo podrán llevar a cabo la actividad prevista en el artículo 5.5 del Reglamento (UE) 2017/745 y cumplir todos los requisitos de control y calidad. No se aplicarán los requisitos del Reglamento, a excepción de los requisitos generales de seguridad y funcionamiento.
a) Que los productos no se cedan a otras personas jurídicas, empresas, hospitales o centros no dependientes de él. No se permitirá la venta al público de productos fabricados en hospitales. Los hospitales no podrán vender ni entregar el producto fabricado en su hospital para su uso por terceros
b) Que la fabricación y uso de los productos tengan lugar en el marco de sistemas de gestión de calidad apropiados como la UNE EN ISO 13485. Este es el proceso:
Sólo he encontrado este artículo que habla específicamente de esterilización y productos 3D. Recomiendan el óxido de etileno, y en el caso de usar gas plasma, dicen que hay que usar un relleno o no utilizarlo en el caso de dispositivos huecos. Hay más artículos sobre diferentes técnicas, materiales, medición de resultados y deformidades; pero desgraciadamente no hay ninguna norma que homogeneice estos estudios ¿debemos crear una UNE-EN ISO sobre el tema?.
c) Que el centro sanitario justifique en su documentación que no pueden satisfacerse las necesidades específicas del grupo de pacientes al que se destinan los productos, o no pueden satisfacerse con el nivel de funcionamiento adecuado, mediante otro producto equivalente comercializado.
d) Que el centro sanitario facilite a su autoridad competente información sobre el uso de dichos productos en la que se incluirá una justificación de su fabricación, modificación y uso.
f) Que el centro sanitario elabore una documentación que haga posible conocer la instalación de producción, el proceso de producción, el diseño y los datos de funcionamiento de los productos, incluida la finalidad prevista.
Además hay una serie de aspectos que conviene recordar:
Los productos de clase IIb, clase III e implantables NO podrán ser objeto de fabricación por los hospitales que desempeñen la actividad prevista en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017. Yo sigo con la duda que ya expresé en mi entrada anterior, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitará una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida pero de productos a medida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Los hospitales no podrán subcontratar ninguna de las actividades de fabricación. ¿la esterilización forma parte de la fabricación?
Se designará una persona responsable para los procedimientos que se comunicará a la Agencia Española de Medicamentos y Productos Sanitarios. Creo que hay muchos preventivistas que no saben este tema.
Parece que la pandemia nos ha hecho aprender algo. No obstante, la Agencia Española de Medicamentos y Productos Sanitarios, de forma excepcional en casos de emergencia sanitaria, podrá autorizar la fabricación de cualquier producto en centros sanitarios o institutos de salud pública en condiciones distintas a las previstas en este artículo, cuando su utilización redunde en beneficio de la salud pública o de la seguridad o salud de los pacientes.
Cada vez hay más experiencias de los hospitales, y muchos papers:
La licencia de productos a medida se basa en el Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para la concesión de licencias de funcionamiento a los fabricantes de productos sanitarios a medida. Para definir lo que es un producto sanitario a medida se va al artículo 2.a). Y este artículo 2.a) dice ««Producto sanitario», «accesorio», «producto a medida», «fabricante» y «comercialización», lo que se establece en el artículo 3 del Real Decreto 414/1996, de 1 de marzo, por el se regula los productos sanitarios». Pero es que el Real Decreto 414/1996 está derogado por el Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios; que está totalmente derogado (excepto una parte) por el Real Decreto 192/2023. Habrá que actualizar la legislación.
Clase I: productos que no entran en contacto con el paciente o que entran en contacto solo con la piel intacta. Productos que penetran por orificio corporal, como la boca o la nariz, de uso pasajero.
Ejemplos: productos para recolección de fluidos corporales (bolsas de orina), productos para inmovilizar partes del cuerpo o para aplicar compresión (vendas, medias elásticas), productos para el apoyo del paciente (andadores, bastones) y otros (gafas, enemas, lámparas de reconocimiento). Se excluyen de esta clase los productos que, aunque no entran en contacto con el paciente, pueden influir en procesos fisiológicos (productos que tratan la sangre destinada a reinfundirse) o los que suministran energía al cuerpo humano (equipos de radiodiagnóstico).
Dentro de la clase I podemos encontrar también:
Clase I estériles. Ejemplos: guantes de examen, jeringuillas, equipos de infusión por gravedad, gasas para proteger las heridas o para absorber exudados, instrumentos quirúrgicos reutilizables.
Clase I con función de medición. Ejemplos: jeringuillas, termómetros no electrónicos, tonómetros.
Clase IIa: se incluyen en esta clase los productos que se introducen en el cuerpo humano por orificio corporal o por medios quirúrgicos, es decir a través de la piel, pero que no están destinados a permanecer en él. También los que suministran energía o sustancias, o los que modifican procesos fisiológicos siempre y cuando no se efectúe de forma potencialmente peligrosa. También se incluyen en esta clase los desinfectantes de productos no invasivos.
Ejemplos: lentillas de contacto, los equipos de ecografía, las coronas dentales; los termómetro, circuitos de circulación extracorpórea, sondas urológicas, drenajes quirúrgicos, agujas, cánulas, guantes quirúrgicos, lentes de contacto, audífonos, estimuladores musculares: TENS, esfigmomanómetros, equipos de diagnóstico, equipos para fisioterapia.
Clase IIb: se incluyen algunos productos implantables (aunque se clasifican muchos de ellos como clase III), los productos que pueden influenciar los procesos fisiológicos o que administran sustancias o energía de forma potencialmente peligrosa y los que se destinan al diagnóstico de funciones vitales. También se clasifican como IIb los productos sanitarios anticonceptivos o para la prevención de enfermedades de transmisión sexual y los desinfectantes de productos invasivos, así como los productos para el cuidado de lentes de contacto.
Ejemplos: preservativos, los productos desinfectantes de las lentillas,lentes intraoculares, implantes de relleno tisular, suturas quirúrgicas no absorbibles, apósitos para heridas que cicatrizan por segunda intención, bolsas de sangre, hemodializadores, plumas de insulina, desfibriladores externos, equipos de rayos X para diagnóstico, láseres quirúrgicos, equipos para terapia por radiaciones, sistemas de vigilancia para cuidados intensivos, máquinas de anestesia, preservativos, etc.
Clase III: se incluyen en esta clase algunos productos implantables, los productos destinados a entrar en contacto con el sistema nervioso central o con el sistema circulatorio central con fines de terapia o diagnóstico, los productos que contienen sustancias medicinales, los productos que se absorben to-talmente y los productos que contienen derivados animales.
Ejemplos: válvulas cardíacas, prótesis de cadera, prótesis de mama, endoprótesis vasculares: stents, catéteres cardiovasculares, suturas absorbibles, adhesivos de tejidos internos biológicos, materiales de endodoncia con antibióticos, apósitos con agentes antimicrobianos.
Y muy importante recordar que los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales.
El video mató a la estrella de la radio, ¿pasará lo mismo con las impresiones 3D y el modo de operar a nuestros pacientes? ¿ha llegado la cirugía personalizada?
Aquí no acaba la entrada del Blog ¿Qué son los productos a medida? (Artículo 10 del RD 192/2023): «todo producto fabricado especialmente según la prescripción médica de cualquier persona autorizada por la legislación nacional en virtud de su cualificación profesional, en la que constan, bajo la responsabilidad de dicha persona, las características específicas de diseño, y que está destinado a ser utilizado únicamente por un paciente determinado con el fin exclusivo de atender a su estado y necesidades particulares» (artículo 2.3 del Reglamento 2017/745).
Para crear estos productos se requiere licencia previa de funcionamiento otorgada por las autoridades sanitarias de la comunidad autónoma correspondiente, de conformidad con el Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para concesión de licencias de funcionamiento para fabricantes de productos a medida. Deberán inscribirse en el Registro de responsables de la puesta en el mercado de productos a medida a través de la sede electrónica de la Agencia Española de Medicamentos y Productos Sanitarios.
Leído esto, los hospitales que impriman productos 3D específicos para un paciente deberán obtener la licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Pero claro, el Reglamento dice que no podrán ser IIb y III.
Anatomical model (CMD CLass I) for surgical planning in case of giant coronary artery bypass aneurysm manufactured with FDM technology
Surgical guide for implantology (Class IIa CMD) 3D printed in biocompatible material
Surgical guide (CMD Class IIb) in case of pectus carinatum. 3D Printed and manufactured in biocompatible material
Tomado del Webinar de la AEMPS 23/05/2023
Tomado del Webinar de la AEMPS 23/05/2023
Al final, lo que importa es ésto «The precision in planning and the perfect adaptation of both the cutting guide and the specific metal implant allowed an open approach through the upper eyelid only, so that the shortened surgical time, the minimal morbidity and the aesthetic and functional recovery of the patient have compensated the effort in the personalized design of our surgical treatment».
Realmente ha llegado la cirugía de precisión y personalizada.
La cantante Cher saben mucho de cirugía de precisión y personalizada, con sus arreglos y prótesis.
Para saber la relación de Miguel Bosé con la entrada de productos de un solo uso, deberéis llegar al final de la entrada. No haré spoiler.
Ha costado, pero ha llegado el nuevo Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. Ya está publicado en el BOE, y ¡es gratis! nadie puede decir que no lo ha leído por no estar accesible. No es magia, son tus impuestos. Nos quejamos de que las normas UNE-EN ISO son de pago, y ahora tenemos 29 folios gratuitos y accesibles, y además te avisan si se cambia algo del reglamento con el paso de tiempo si estas suscrito a «Mi BOE».
Este Real Decreto viene a esclarecer algunas cosas que no había dejado escrito el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, y del que ya hablé en este Blog.
Para no aburriros, lo que voy a hacer son pequeñas entradas de lo que me parece más importante que viene en este Real Decreto (Reglamento) que es de obligado cumplimiento en todo el territorio español (esto no entiende de autonomías) desde el 23 de marzo de 2023, día de San José Oriol. Haré entradas relativas a (este orden no es el de aparición en el Blog, si no que sigo el del reglamento):
Definiciones y aspectos a tener en cuenta (Capítulo I del Real Decreto).
Las Licencias de Funcionamiento (Capítulo II), que parece que sólo se va a pedir a las empresas que se dedican a reprocesar (empresas subcontratadas o externalizadas) o cuando un hospital sirve un producto a un hospital que no es de su grupo hospitalario (como los privados) o de su área sanitaria o un centro de centro de salud que no es de gerencia o área. Por supuesto, no puede esterilizar algo y dárselo a una clínica privada. Las licencias de funcionamiento serán necesarias en instalaciones a fabricantes, esterilizadores, agrupadores e importadores de producto sanitario y las instalaciones en que se lleven a cabo dichas actividades. Este requisito será también de aplicación a empresas que realicen un reprocesamiento de producto sanitario de un solo uso, fabricantes de productos sanitarios acogidos en el anexo XVI, aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas y la fabricación completa de los productos para terceros.
Fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II). ¿Quién no ha recibido ya alguna prótesis o producto metido en una bolsa con un folio y que se lo esterilicen? No se puede hacer si no es con unas condiciones que ya explicaré. Lo principal es que deberán realizar una comunicación previa, los `productos fabricados y utilizados en el mismo centro sanitario, los hospitales no podrán subcontratar ninguna de las actividades de fabricación, que no existan alternativas en el mercado. Y muy importante los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales. Y aquí viene algo que me llama la atención, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitar una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Reprocesado de productos de un solo uso (Capítulo III), que es de lo que hablaré hoy.
El Capítulo IV os interesa menos porque habla del Organismo notificado. Pero el Capítulo V si que interesa por que es el que trata la trazabilidad.
Y nos quedarían el VI, VII, VIII y IX que haré un resumen.
Es importante decir que las definiciones de este reglamento ya venían en el reglamento europeo. A los efectos de este real decreto, se aplicarán las definiciones recogidas en el artículo 2 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios; por lo que no hay cambios. Pero me parece importante indicar que se incluyen:
Los aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas (Art. 3.1.b). Los tatuadores y maquilladores deberán cumplir las mismas normas que la central en cuanto a seguridad. Algo hablé del tema cuando hice la entrada del brote de tiña en las peluquerías y el virus del Monkeypox.
Cuando un producto destinado por su fabricante a ser utilizado como equipo de protección individual esté destinado también a ser utilizado como producto sanitario deberá cumplir, además de la normativa aplicable a los productos de protección individual, las disposiciones de este real decreto (Art. 3.2).
La gran novedad de este reglamento es que ya se permite reprocesar productos de un solo uso. Y ya no podré cantar la canción de Camilo Sesto que tanto me gusta.
Diapositiva de una de mis ponencias
Lo he repetido mucho en este Blog que estaba prohibido reprocesa productos de un solo uso:
Diapositiva del Dr Luis Salmerón, basada en mi Blog
Y lo dice el título del Capítulo III «Reprocesamiento y nueva utilización de productos de un solo uso», habla del reprocesamiento y nueva utilización. Porque incido en lo de nueva utilización, por que mucha gente que me ha oído en foros, siempre hablo de la funcionalidad de los preservativos caducados en las carteras de los adolescentes de los años 80. Ahora tenemos que demostrar que el producto que reprocesamos funciona como si fuera nuevo. Eso no lo podemos hacer con los medios de nuestros hospitales.
En el año 2010 la Comisión Europea elaboró el Informe sobre el reprocesamiento de productos sanitarios en la Unión Europea que dice «Contrariamente a los productos sanitarios reutilizables, para los cuales se establecen requisitos en la Directiva 93/42/CEE a fin de garantizar una reutilización segura, en el caso de los productos sanitarios de un solo uso no puede afirmarse que su reutilización no presente riesgos desde el punto de vista de la salud pública. Además, también deberían tenerse en cuenta los aspectos éticos, económicos, medioambientales y relacionados con la responsabilidad del reprocesamiento de productos sanitarios de un solo uso, por lo que se analizan en mayor detalle en el presente informe».
«Los productos sanitarios de un solo uso, tales como agujas o catéteres de angioplastia, no están concebidos y diseñados para resistir a un procedimiento de reprocesamiento y el fabricante no necesita proporcionar ninguna instrucción o procedimiento validado para permitir un reprocesamiento seguro del producto, sino solamente información sobre las características o los factores técnicos conocidos por el fabricante que podrían presentar un riesgo en caso de que se reutilizara el producto. Por consiguiente, el reprocesamiento se realiza a partir de procedimientos elaborados por el usuario o el prestatario de servicios de reprocesamiento, pero sin información completa sobre el diseño y la composición del producto. Según un informe de los Países Bajos[16], la validación de un procedimiento de reprocesamiento para productos sanitarios de un solo uso, especialmente la limpieza, es una tarea que normalmente no puede realizarse en un hospital, ya que es poco probable que se disponga del equipo, el conocimiento, la experiencia y los recursos requeridos».
«No todos los productos sanitarios de un solo uso son adecuados para ser reprocesados debido a sus características o a la complejidad de determinados de estos productos. La posibilidad de reprocesamiento depende del material utilizado y de la forma del producto sanitario. Con el fin de identificar y reducir los peligros potenciales asociados con el reprocesamiento de un producto sanitario de un solo uso específico, debe evaluarse y validarse todo el ciclo de reprocesamiento que se inicia con la recogida de estos productos sanitarios de un solo uso después de su primera utilización hasta la fase final de esterilización y entrega, incluidas sus prestaciones funcionales.»
Así que el Reglamento permite reprocesa productos de un solo uso, pero NO SIGNIFICA BARRA LIBRE. Se puede hacer, pero siguiendo unas estrictas normas de calidad y seguridad «podrán llevarse a cabo siempre y cuando se cumplan los requisitos del presente real decreto» (Art. 11.1).
La gestión de riesgos: Tiene que existir un sistema. Para eso deberemos certificarnos (que es diferente a autorizarse y acreditarse) mediante alguna UNE-EN ISO como la UNE 179003 y la UNE-EN ISO 14971 (lo siento son de pago y no las puedo poner en el Blog, por que me llevarían a la cárcel). No quiero ser como Alexandra Elbakyan y su preciado sci-hub ¿Quién no lo ha utilizado alguna vez?
Un sistema de gestión de la calidad como la UNE-EN ISO 13485 (que es la adaptación al producto sanitario de la 9001).
En este tema de las UNE-EN ISO siempre puede ayudarnos (previo pago) una empresa certificadora, que generalmente les enseñas todo, les haces el trabajo y ellos dicen que haces las cosas como las describes en el manual (¡Viva los notarios del reino!). Ya sabéis lo poco que me gustan estos sistemas de auditoría por estas empresas con ánimo de lucro (incesante). Estos organismos que nos certifican deben realizarnos auditorías anuales (organismos acreditados para la certificación de sistemas de calidad de productos sanitarios) (Art. 13.5).
Tener estos dos sistema de calidad y gestión de riesgos nos obliga a validar los equipos de limpieza y termodesinfección, las selladoras y los autoclaves (frío y vapor), además de los residuos que se producen en los dispositivos médicos. TODOS. Y las validaciones cuestan mucho dinero y esfuerzo.
Trazabilidad: Eso al menos ya lo hacemos la gran mayoría de centrales o RUMED.
Notificar incidentes.
Y deberemos asegurarnos que funciona igual que antes. La funcionalidad es muy importante, y demostrarla muy difícil.
En conclusión. Los hospitales solo podrán reprocesar productos que hayan sido utilizados y reprocesados en su hospital o por un reprocesador externo incluido en su licencia. Los hospitales no podrán vender ni entregar el producto reprocesado a terceros (Art. 13.2 y 13.3).
Los hospitales podrán subcontratar las actividades de reprocesamiento a un reprocesador externo (Art. 14). OJO con estas empresas, ya que deben tener su domicilio social e instalaciones establecidas en España. Vamos que no se puede reprocesar en China o Pakistán (por ejemplo), si no que deben tener su domicilio en algún lugar del territorio español (por ejemplo en Quismondo o Pelahustán, que no son repúblicas exsocialistas soviéticas. Existen). Los reprocesadores externos deberán cumplir toda la normativa y no pueden subcontratar las actividades de reprocesamiento.
Y hay dos hechos que me parecen novedosos (Art. 15):
«Los hospitales deberán informar a los pacientes de la utilización en su hospital de productos reprocesados por su propio centro». A partir de ahora se debe informar al paciente y reflejarlo en su historia clínica.
Los productos reprocesados únicamente podrán utilizarse en los hospitales en un único paciente y durante un único proceso. Ya no vale reprocesar continuamente y a lo loco.
Una de las ventajas de permitir el reprocesado de productos de un un solo uso, sería reducir la huella de carbono de nuestros hospitales. Es un tema estudiado con los catéteres cardiológicos y catéteres de ablación, o intervenciones como las cataratas.
Cada vez serán más frecuentes los estudios de huella de carbono en hospitales o el bloque quirúrgico, endoscopias, y estudiar maneras de reducirla como la reutilización, reuso y reciclado. Quizás el medioambiente sea un motivo (y muy importante) de reprocesar productos de un solo uso .
Hay una serie de productos sanitarios que no se podrán reprocesar (Art. 11.2):
a) De clase I. Por ejemplo, bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas, guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas, tonómetros o termómetros no electrónicos.
b) A medida. OJO CON ÉSTOS
c) Fabricados y utilizados exclusivamente en hospitales de acuerdo con lo establecido en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017.
d) Que emitan radiación.
e) Utilizados para la administración de medicamentos citostáticos o radiofármacos.
f) Que incorporen sustancias medicinales.
g) Para uso en procedimientos invasivos en el sistema nervioso central.
h) Que presenten un riesgo de transmisión de encefalopatías espongiformes.
i) Implantables. OJO CON ÉSTOS, se definen como todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina aser introducido totalmente en el cuerpo humano osustituir una superficie epitelial o la superficie ocular,mediante intervención médica, y a permanecer en su lugar después de la intervención.Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
j) Relacionados con incidentes graves ocurridos tras el reprocesamiento cuya causa esté relacionada con el reprocesamiento, o para los que no pueda excluirse que la causa esté relacionada con el reprocesamiento.
k) Que tengan baterías que no puedan cambiarse o que presenten un riesgo de mal funcionamiento tras el reprocesamiento.
l) Que dispongan de un almacenamiento interno de datos necesario para el uso del producto y que no pueda cambiarse o presente un riesgo de mal funcionamiento tras el reprocesamiento.
m) Con hojas cortantes o que raspen, taladros o componentes que se desgasten que dejen de ser adecuados después del primer uso y que no puedan cambiarse o afilarse antes del siguiente procedimiento médico. OJO CON ÉSTOS, ¿COMO SABEMOS QUE LAS HOJAS NO ESTÁN AFILADAS EN UN HOSPITAL?
Así que ya sabéis, si os traen algo a reprocesar que es de un solo uso, o que se ha caducado en el almacén, o que se ha abierto por error y no se ha usado, SOLO PODÉIS REPROCESARLO SI TENÉIS LO INDICADO MÁS ARRIBA.
Y ya no es como antes, que el cirujano o traumatólogo de turno (en plan torero) te firmaba un documento, y se hacía responsable o que tu transferías la responsabilidad. Eso se ha acabado.
Para saber la relación de Miguel Bosé con la entrada de productos de un solo uso, deberéis llegar al final de la entrada. No haré spoiler.
Ha costado, pero ha llegado el nuevo Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. Ya está publicado en el BOE, y ¡es gratis! nadie puede decir que no lo ha leído por no estar accesible. No es magia, son tus impuestos. Nos quejamos de que las normas UNE-EN ISO son de pago, y ahora tenemos 29 folios gratuitos y accesibles, y además te avisan si se cambia algo del reglamento con el paso de tiempo si estas suscrito a «Mi BOE».
Este Real Decreto viene a esclarecer algunas cosas que no había dejado escrito el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, y del que ya hablé en este Blog.
Para no aburriros, lo que voy a hacer son pequeñas entradas de lo que me parece más importante que viene en este Real Decreto (Reglamento) que es de obligado cumplimiento en todo el territorio español (esto no entiende de autonomías) desde el 23 de marzo de 2023, día de San José Oriol. Haré entradas relativas a (este orden no es el de aparición en el Blog, si no que sigo el del reglamento):
Definiciones y aspectos a tener en cuenta (Capítulo I del Real Decreto).
Las Licencias de Funcionamiento (Capítulo II), que parece que sólo se va a pedir a las empresas que se dedican a reprocesar (empresas subcontratadas o externalizadas) o cuando un hospital sirve un producto a un hospital que no es de su grupo hospitalario (como los privados) o de su área sanitaria o un centro de centro de salud que no es de gerencia o área. Por supuesto, no puede esterilizar algo y dárselo a una clínica privada. Las licencias de funcionamiento serán necesarias en instalaciones a fabricantes, esterilizadores, agrupadores e importadores de producto sanitario y las instalaciones en que se lleven a cabo dichas actividades. Este requisito será también de aplicación a empresas que realicen un reprocesamiento de producto sanitario de un solo uso, fabricantes de productos sanitarios acogidos en el anexo XVI, aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas y la fabricación completa de los productos para terceros.
Fabricación por los hospitales de productos para su propio y exclusivo uso (Capítulo II) y los productos a medida (Capítulo II). ¿Quién no ha recibido ya alguna prótesis o producto metido en una bolsa con un folio y que se lo esterilicen? No se puede hacer si no es con unas condiciones que ya explicaré. Lo principal es que deberán realizar una comunicación previa, los `productos fabricados y utilizados en el mismo centro sanitario, los hospitales no podrán subcontratar ninguna de las actividades de fabricación, que no existan alternativas en el mercado. Y muy importante los productos de clase IIb, clase III e implantables no podrán ser objeto de fabricación por los hospitales. Y aquí viene algo que me llama la atención, tú puedes fabricar un producto de clase III en el hospital, pero si se considera producto a medida, el hospital solicitar una licencia previa de funcionamiento que será otorgada por las autoridades sanitarias de la Comunidad Autónoma donde resida. Esto me lo tienen que aclarar ¿podemos fabricar un IIb o III de resina y esterilizarlo en un hospital con una licencia de producto a medida?.
Reprocesado de productos de un solo uso (Capítulo III), que es de lo que hablaré hoy.
El Capítulo IV os interesa menos porque habla del Organismo notificado. Pero el Capítulo V si que interesa por que es el que trata la trazabilidad.
Y nos quedarían el VI, VII, VIII y IX que haré un resumen.
Es importante decir que las definiciones de este reglamento ya venían en el reglamento europeo. A los efectos de este real decreto, se aplicarán las definiciones recogidas en el artículo 2 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios; por lo que no hay cambios. Pero me parece importante indicar que se incluyen:
Los aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas (Art. 3.1.b). Los tatuadores y maquilladores deberán cumplir las mismas normas que la central en cuanto a seguridad. Algo hablé del tema cuando hice la entrada del brote de tiña en las peluquerías y el virus del Monkeypox.
Cuando un producto destinado por su fabricante a ser utilizado como equipo de protección individual esté destinado también a ser utilizado como producto sanitario deberá cumplir, además de la normativa aplicable a los productos de protección individual, las disposiciones de este real decreto (Art. 3.2).
La gran novedad de este reglamento es que ya se permite reprocesar productos de un solo uso. Y ya no podré cantar la canción de Camilo Sesto que tanto me gusta.
Diapositiva de una de mis ponencias
Lo he repetido mucho en este Blog que estaba prohibido reprocesa productos de un solo uso:
Diapositiva del Dr Luis Salmerón, basada en mi Blog
Y lo dice el título del Capítulo III «Reprocesamiento y nueva utilización de productos de un solo uso», habla del reprocesamiento y nueva utilización. Porque incido en lo de nueva utilización, por que mucha gente que me ha oído en foros, siempre hablo de la funcionalidad de los preservativos caducados en las carteras de los adolescentes de los años 80. Ahora tenemos que demostrar que el producto que reprocesamos funciona como si fuera nuevo. Eso no lo podemos hacer con los medios de nuestros hospitales.
En el año 2010 la Comisión Europea elaboró el Informe sobre el reprocesamiento de productos sanitarios en la Unión Europea que dice «Contrariamente a los productos sanitarios reutilizables, para los cuales se establecen requisitos en la Directiva 93/42/CEE a fin de garantizar una reutilización segura, en el caso de los productos sanitarios de un solo uso no puede afirmarse que su reutilización no presente riesgos desde el punto de vista de la salud pública. Además, también deberían tenerse en cuenta los aspectos éticos, económicos, medioambientales y relacionados con la responsabilidad del reprocesamiento de productos sanitarios de un solo uso, por lo que se analizan en mayor detalle en el presente informe».
«Los productos sanitarios de un solo uso, tales como agujas o catéteres de angioplastia, no están concebidos y diseñados para resistir a un procedimiento de reprocesamiento y el fabricante no necesita proporcionar ninguna instrucción o procedimiento validado para permitir un reprocesamiento seguro del producto, sino solamente información sobre las características o los factores técnicos conocidos por el fabricante que podrían presentar un riesgo en caso de que se reutilizara el producto. Por consiguiente, el reprocesamiento se realiza a partir de procedimientos elaborados por el usuario o el prestatario de servicios de reprocesamiento, pero sin información completa sobre el diseño y la composición del producto. Según un informe de los Países Bajos[16], la validación de un procedimiento de reprocesamiento para productos sanitarios de un solo uso, especialmente la limpieza, es una tarea que normalmente no puede realizarse en un hospital, ya que es poco probable que se disponga del equipo, el conocimiento, la experiencia y los recursos requeridos».
«No todos los productos sanitarios de un solo uso son adecuados para ser reprocesados debido a sus características o a la complejidad de determinados de estos productos. La posibilidad de reprocesamiento depende del material utilizado y de la forma del producto sanitario. Con el fin de identificar y reducir los peligros potenciales asociados con el reprocesamiento de un producto sanitario de un solo uso específico, debe evaluarse y validarse todo el ciclo de reprocesamiento que se inicia con la recogida de estos productos sanitarios de un solo uso después de su primera utilización hasta la fase final de esterilización y entrega, incluidas sus prestaciones funcionales.»
Así que el Reglamento permite reprocesa productos de un solo uso, pero NO SIGNIFICA BARRA LIBRE. Se puede hacer, pero siguiendo unas estrictas normas de calidad y seguridad «podrán llevarse a cabo siempre y cuando se cumplan los requisitos del presente real decreto» (Art. 11.1).
La gestión de riesgos: Tiene que existir un sistema. Para eso deberemos certificarnos (que es diferente a autorizarse y acreditarse) mediante alguna UNE-EN ISO como la UNE 179003 y la UNE-EN ISO 14971 (lo siento son de pago y no las puedo poner en el Blog, por que me llevarían a la cárcel). No quiero ser como Alexandra Elbakyan y su preciado sci-hub ¿Quién no lo ha utilizado alguna vez?
Un sistema de gestión de la calidad como la UNE-EN ISO 13485 (que es la adaptación al producto sanitario de la 9001).
En este tema de las UNE-EN ISO siempre puede ayudarnos (previo pago) una empresa certificadora, que generalmente les enseñas todo, les haces el trabajo y ellos dicen que haces las cosas como las describes en el manual (¡Viva los notarios del reino!). Ya sabéis lo poco que me gustan estos sistemas de auditoría por estas empresas con ánimo de lucro (incesante). Estos organismos que nos certifican deben realizarnos auditorías anuales (organismos acreditados para la certificación de sistemas de calidad de productos sanitarios) (Art. 13.5).
Tener estos dos sistema de calidad y gestión de riesgos nos obliga a validar los equipos de limpieza y termodesinfección, las selladoras y los autoclaves (frío y vapor), además de los residuos que se producen en los dispositivos médicos. TODOS. Y las validaciones cuestan mucho dinero y esfuerzo.
Trazabilidad: Eso al menos ya lo hacemos la gran mayoría de centrales o RUMED.
Notificar incidentes.
Y deberemos asegurarnos que funciona igual que antes. La funcionalidad es muy importante, y demostrarla muy difícil.
En conclusión. Los hospitales solo podrán reprocesar productos que hayan sido utilizados y reprocesados en su hospital o por un reprocesador externo incluido en su licencia. Los hospitales no podrán vender ni entregar el producto reprocesado a terceros (Art. 13.2 y 13.3).
Los hospitales podrán subcontratar las actividades de reprocesamiento a un reprocesador externo (Art. 14). OJO con estas empresas, ya que deben tener su domicilio social e instalaciones establecidas en España. Vamos que no se puede reprocesar en China o Pakistán (por ejemplo), si no que deben tener su domicilio en algún lugar del territorio español (por ejemplo en Quismondo o Pelahustán, que no son repúblicas exsocialistas soviéticas. Existen). Los reprocesadores externos deberán cumplir toda la normativa y no pueden subcontratar las actividades de reprocesamiento.
Y hay dos hechos que me parecen novedosos (Art. 15):
«Los hospitales deberán informar a los pacientes de la utilización en su hospital de productos reprocesados por su propio centro». A partir de ahora se debe informar al paciente y reflejarlo en su historia clínica.
Los productos reprocesados únicamente podrán utilizarse en los hospitales en un único paciente y durante un único proceso. Ya no vale reprocesar continuamente y a lo loco.
Una de las ventajas de permitir el reprocesado de productos de un un solo uso, sería reducir la huella de carbono de nuestros hospitales. Es un tema estudiado con los catéteres cardiológicos y catéteres de ablación, o intervenciones como las cataratas.
Cada vez serán más frecuentes los estudios de huella de carbono en hospitales o el bloque quirúrgico, endoscopias, y estudiar maneras de reducirla como la reutilización, reuso y reciclado. Quizás el medioambiente sea un motivo (y muy importante) de reprocesar productos de un solo uso .
Hay una serie de productos sanitarios que no se podrán reprocesar (Art. 11.2):
a) De clase I. Por ejemplo, bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas, guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas, tonómetros o termómetros no electrónicos.
b) A medida. OJO CON ÉSTOS
c) Fabricados y utilizados exclusivamente en hospitales de acuerdo con lo establecido en el artículo 5.5 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017.
d) Que emitan radiación.
e) Utilizados para la administración de medicamentos citostáticos o radiofármacos.
f) Que incorporen sustancias medicinales.
g) Para uso en procedimientos invasivos en el sistema nervioso central.
h) Que presenten un riesgo de transmisión de encefalopatías espongiformes.
i) Implantables. OJO CON ÉSTOS, se definen como todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina aser introducido totalmente en el cuerpo humano osustituir una superficie epitelial o la superficie ocular,mediante intervención médica, y a permanecer en su lugar después de la intervención.Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
j) Relacionados con incidentes graves ocurridos tras el reprocesamiento cuya causa esté relacionada con el reprocesamiento, o para los que no pueda excluirse que la causa esté relacionada con el reprocesamiento.
k) Que tengan baterías que no puedan cambiarse o que presenten un riesgo de mal funcionamiento tras el reprocesamiento.
l) Que dispongan de un almacenamiento interno de datos necesario para el uso del producto y que no pueda cambiarse o presente un riesgo de mal funcionamiento tras el reprocesamiento.
m) Con hojas cortantes o que raspen, taladros o componentes que se desgasten que dejen de ser adecuados después del primer uso y que no puedan cambiarse o afilarse antes del siguiente procedimiento médico. OJO CON ÉSTOS, ¿COMO SABEMOS QUE LAS HOJAS NO ESTÁN AFILADAS EN UN HOSPITAL?
Así que ya sabéis, si os traen algo a reprocesar que es de un solo uso, o que se ha caducado en el almacén, o que se ha abierto por error y no se ha usado, SOLO PODÉIS REPROCESARLO SI TENÉIS LO INDICADO MÁS ARRIBA.
Y ya no es como antes, que el cirujano o traumatólogo de turno (en plan torero) te firmaba un documento, y se hacía responsable o que tu transferías la responsabilidad. Eso se ha acabado.
Una vez más, las enfermeras quirúrgicas tendrán la oportunidad de poner en común sus conocimientos y proyectos de investigación enfermera. También podrán compartir experiencias profesionales que permitan contribuir a una sanidad más eficiente, más segura y cercana al paciente. Sin duda una experiencia enriquecedora que como enfermeras quirúrgicas no os podéis perder.
Organizadas por la Asociación Española de Enfermería Quirúrgica (AEEQ), los comités están preparando un interesante e innovador programa científico que abordará diversas áreas de la enfermería quirúrgica. Se analizarán y profundizaremos en temas como: las nuevas tecnologías, la investigación enfermera, la legislación o la esterilización.
El programa científico incluirá conferencias magistrales, mesas redondas y symposium, además de diferentes talleres eminentemente prácticos en los que asistentes profundizarán en las distintas especialidades quirúrgicas. Cabe destacar el elenco de ponentes que participarán, expertos en cada uno de los temas que se abordarán durante las sesiones científicas.
También tenéis el hashtag #17AEEQ, para que podáis utilizarla en vuestras redes sociales.
Participaré en este congreso con la ponencia «First dates ¿Tendrías una segunda cita con Esterilización?».
La central de esterilización y el bloque quirúrgico son como un «matrimonio», hay días en que las cosas van muy bien y otras en que es mejor no decirse nada. Como Pimpinela. Si voy bien de tiempo en mi presentación en el congreso, intentaré cantar algo de esta pareja. Espero que alguien me acompañe.




























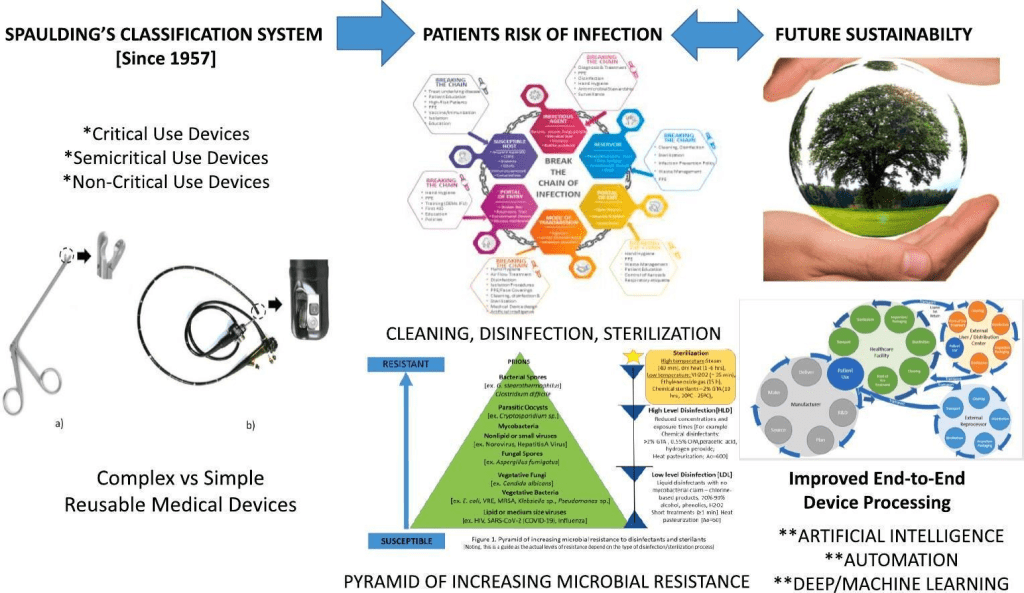

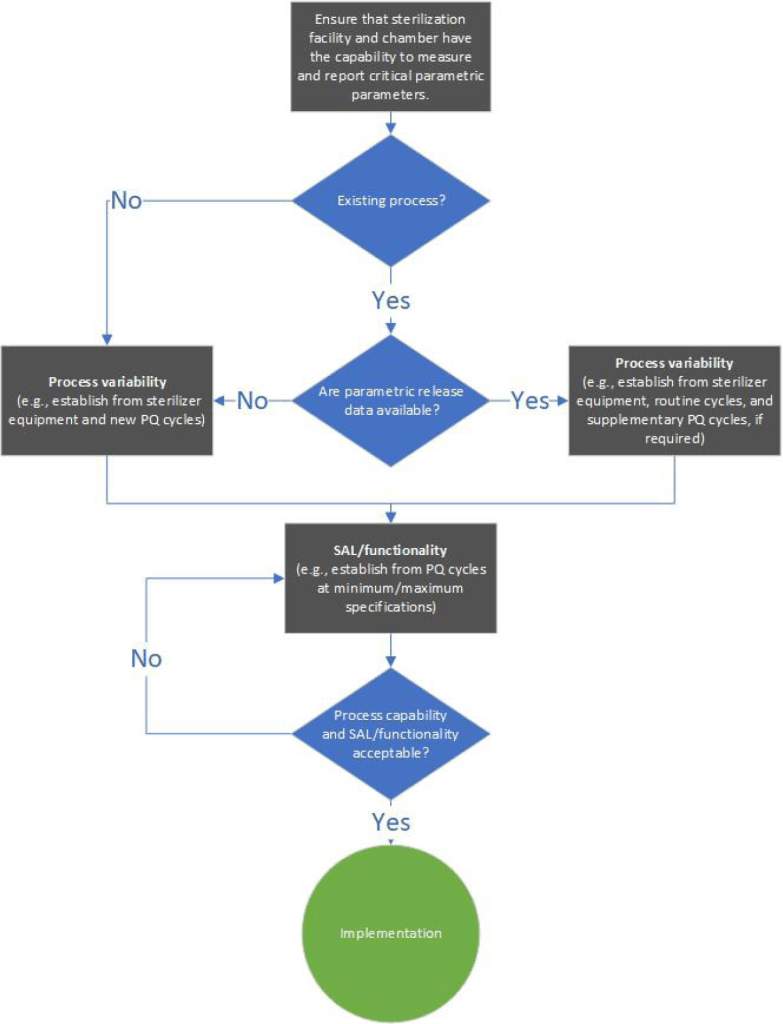










































Debe estar conectado para enviar un comentario.