El blog de la limpieza, desinfección y esterilizacion de dispositivos sanitarios. Este Blog no pertenece ni representa a ninguna Sociedad Científica, Asociación u Organismo, su finalidad es la difusión de conocimientos y actividades relacionados con la Esterilización. Todo es fruto de una búsqueda personal de evidencia en este campo sanitario. El administrador de este blog no se responsibiliza de la información contenida en el blog pues pudieran existir errores de intepretación o traducción en algún caso de los artículos o fuentes originales. Se recomienda, por tanto, consultar con los escritos originales (enlaces), de los que tampoco este administrador se responsabiliza de su exactitud. Tampoco se responsabiliza de las opiniones vertidas por sus seguidores. Los contenidos patrocinados se indicarán debidamente.
Con mucha ilusión están organizando la VIII Jornada del Experto de Socinorte que se celebrará en la ciudad de Tudela (Navarra) el día 27 de mayo de 2022. Los compañeros de SOCINORTE han organizado un buen programa formativo que esperan sea de interés para todos.
Muchas cosas nos han pasado estos dos estos últimos años y nuestra respuesta es “Seguir Adelante”. Que mejor forma de hacerlo que juntándonos en esta pequeña ciudad, llena de historia, para dar continuidad a nuestras reuniones científicas, foro de conocimiento y de compartir experiencias profesionales.
La información científica disponible y la situación pandémica actual ponen de manifiesto, de forma visible y relevante, el importante papel que juega el medio ambiente sanitario y los dispositivos médicos en la transmisión de infecciones relacionadas con la asistencia sanitaria, así como la importancia de la regulación y normalización de los productos y materiales.
Os animamos a que os inscribáis a estas Jornadas gratuitas y nos enviéis vuestras preguntas al experto.
PROGRAMA
09:00 h. Presentación
09:40 h. PANEL DE EXPERTOS
Modera: Montserrat Torres Berdonces. Enfermera de M. Preventiva y Gestión de Calidad Hospital Reina Sofía
09:45 h. Normativa y regulación aplicable a los desinfectantes y biocidas de uso hospitalario. María Aláez. Directora Técnica. Federación Española de Empresas de Tecnología Sanitaria (FENIN)
10:20 h. Seguridad del Paciente y Endoscopia: Prevención de Infección en el reprocesamiento de dispositivos reutilizables. Carmen Martínez Ortega. Especialista en Medicina Preventiva y Salud Pública. Responsable de Servicio de Medicina Preventiva y Salud Pública Hospital Valle del Nalón
11:00 h. Café
11:30 h. Desinfección no touch: ¿Es suficiente la limpieza manual o el frotar se va a acabar?Juan José Criado Álvarez. Director Gerente del Instituto de Ciencias de la Salud de Castilla-La Mancha. Autor del Blog “El autoclave”
12:10 h. Instrumental en tránsito: Calidad y seguridad en su gestión y reprocesamiento. Rosa Orta Álava. Enfermera Área de Salud de Tudela. Experiencia en Gestión del Área quirúrgica del Hospital Reina Sofía. Tudela.
12:50 h. PREGUNTAS AL EXPERTO
Modera: Ingrid Estévez Coro. Especialista en Medicina Preventiva y Salud Pública Responsable de Servicio de Medicina Preventiva y Gestión de Calidad del Hospital Reina Sofía Tudela
13:50 h. Conclusiones de Jornada. Judith Chamorro Camazón. Especialista en Medicina Preventiva y Salud Pública. Jefa de Servicio de Medicina Preventiva e Higiene hospitalaria. Hospital Universitario de Navarra
14:00 h. Clausura. Enrique Peiró Callizo. Presidente de Socinorte. Coordinación de Programas de Salud Pública y de Seguridad del Paciente. Dirección de Asistencia Sanitaria. Osakidetza
Y la despedida con música muy sexy e irresistible (me encanta Rober Palmer). Comparen las estéticas de los dos últimos videos
Es un día de revindicar una profesión. Ya lo hice el año pasado, y me repito éste. Quizás ya ha llegado el momento de exigir la profesionalización de los Técnicos. Hay un proyecto de Real Decreto de «Técnico en Cuidados sanitarios»; que lleva desde abril del 2019 en algún lugar del Ministerio de Educación (aunque el último borrador es de diciembre de 2019) . Pero claro, hemos tenido varios gobiernos, gobiernos en funciones y varias votaciones; y después llegó el Coronavirus. Por que la competencia es del Ministerio de Educación y Formación Profesional; mientras que el Ministerio de Sanidad tan sólo se encarga de las ordenación de las profesiones sanitarias, una vez creadas las especialidades y curriculums.
Vemos que en sus páginas 32 a 37 hay una parte importante de asepsia y antisepsia, limpieza, desinfección y esterilización. Y lo más importante, es que incide en la importancia de esta profesión fuera del hospital. Eso que llamamos los pequeños usuarios y que incluye desde centros de atención primaria (en la pública) a centros médicos privados, odontólogos, podólogos, centros de estética y tatuajes…
Hoy es un día de fiesta y reivindicación.
Pues parece que el 23 Congreso de la WFHSS será en Barcelona.
No a la guerra. Nuestro apoyo a los ciudadanos de Ucrania
«La que se avecina» es el título de la ponencia sobre el nuevo Reglamento, pero la geopolítica se ha colado y también podemos aplicarlo.
Hace unos días «tomé un café» en una sesión organizada por la casa comercial Dr. Weigert sobre el Reglamento 2017/745 (link de la noticia), del que ya hablé en el Blog en una entrada, pero esta vez lo voy a ampliar.
Lugar del encuentro
Me acompañó una amiga y profesional como Mercedes García Haro, que se centró en la aplicación directa en la RUMED. Fue de esas sesiones con amigos, en un ambiente muy distendido y agradable. Una organización inmejorable con el equipo de Dr. Weigert.
Aquí Mercedes iniciando la exposición
¿Qué debo hacer con el nuevo reglamento de productos sanitarios? (así se llamaba la charla). Leerlo y padecerlo en la intimidad como unas hemorroides.
Y aquí empezando mi charla
Este reglamento tiene como objetivo garantizar la disponibilidad en el mercado de productos sanitarios eficaces, de calidad y seguros.
De dónde venimos, y adonde vamos
Reglamento de Productos Sanitarios o Medical Devices Regulation (MDR), son dos conceptos: Reglamento y Productos sanitarios (Artículo 2 del Reglamento 745/2017).
Los reglamentos son actos jurídicos que se aplican de manera automática y uniforme en todos los países de la UE desde su entrada en vigor, sin necesidad de incorporación al derecho nacional.
Son obligatorios, en todos sus elementos, en los Estados miembros. El Reglamento comunitario una norma de aplicación directa, el RD 1591/2009 ha pasado a quedar, en aquellos aspectos que no resulten conformes con aquel, desplazados (que no derogados). El Reglamento es una norma de aplicación directa, pero se supedita algunos aspectos o cuestiones a la regulación que se establezca a nivel nacional. El Reglamento obliga a:
1.- Derogar el Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios (excepto 21, 38, 39 y 40, y el Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos (excepto 18, 34, 35 y 36), ante la aplicación directa del Reglamento (UE) 2017/745.
2.- Desarrollar las medidas reglamentarias necesarias para aquellos aspectos en los que el reglamento comunitario ha determinado que serán los Estados miembros lo que establecerán la regulación a nivel nacional.
3.- Adaptar, adoptar o mantener las medidas requeridas por la legislación nacional.
El futuro Real Decreto es necesario para establecer:
a) Los requisitos y procedimientos para la regulación de los productos fabricados y utilizados en un centro sanitario (fabricación in house)
b) Los requisitos y procedimientos para la regulación del reprocesamiento de productos sanitarios de un solo uso
c) La regulación de la tarjeta de implantación
d) La creación de un registro nacional de comercialización de productos sanitarios
e) Establecer que la autoridad competente es la Agencia Española de Medicamentos y Productos Sanitarios independientemente de las competencias de otras autoridades sanitarias
Según el proyecto de Real Decreto: Todas las centrales de esterilización o RUMED de los hospitales (públicos y privados) tendrán que obtener la Licencia de Funcionamiento.
¿Qué es un producto sanitario?Producto sanitario (Medical Devices: MD) es cualquier instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en seres humanos y que no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.
También se considerarán productos sanitarios los productos de control o apoyo a la concepción. Y atención, los productos destinados específicamente a la limpieza, desinfección y esterilización de los productos sanitarios. Se excluyen las lentes de contacto y productos para introducirse o colocarse en los ojos, y los productos invasivos quirúrgicos para modificar anatomía o fijación partes del cuerpo, excluidos tatuajes y piercings.
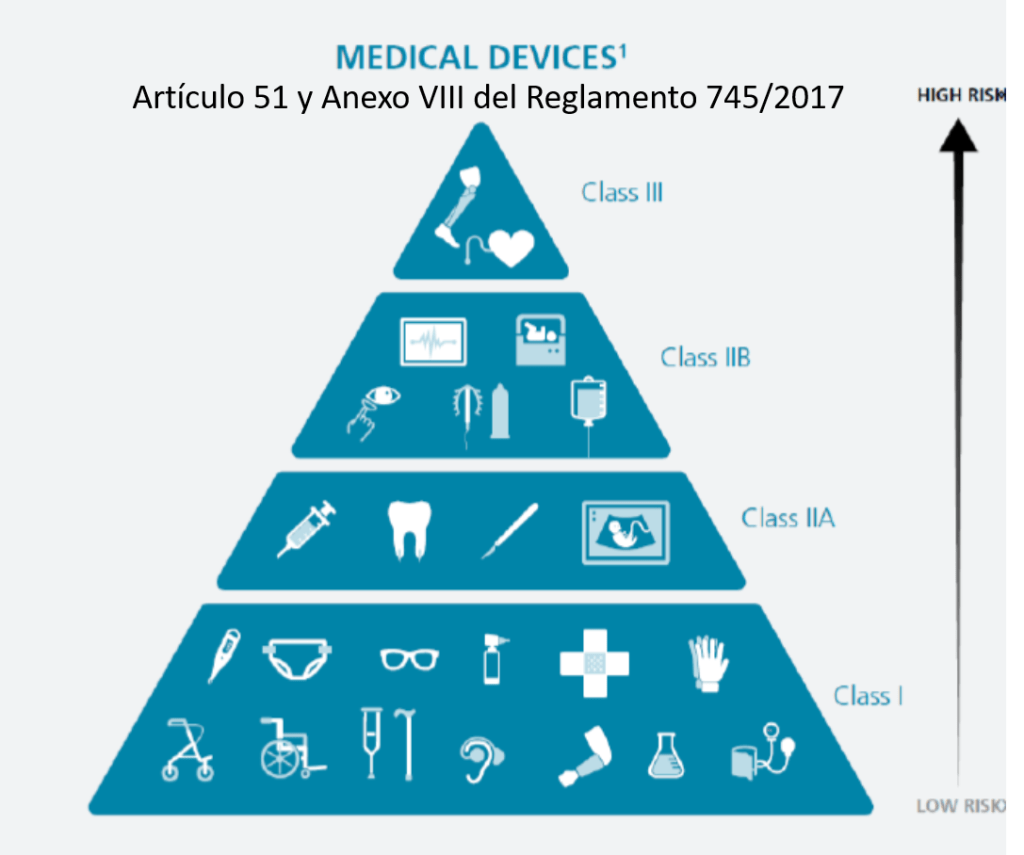
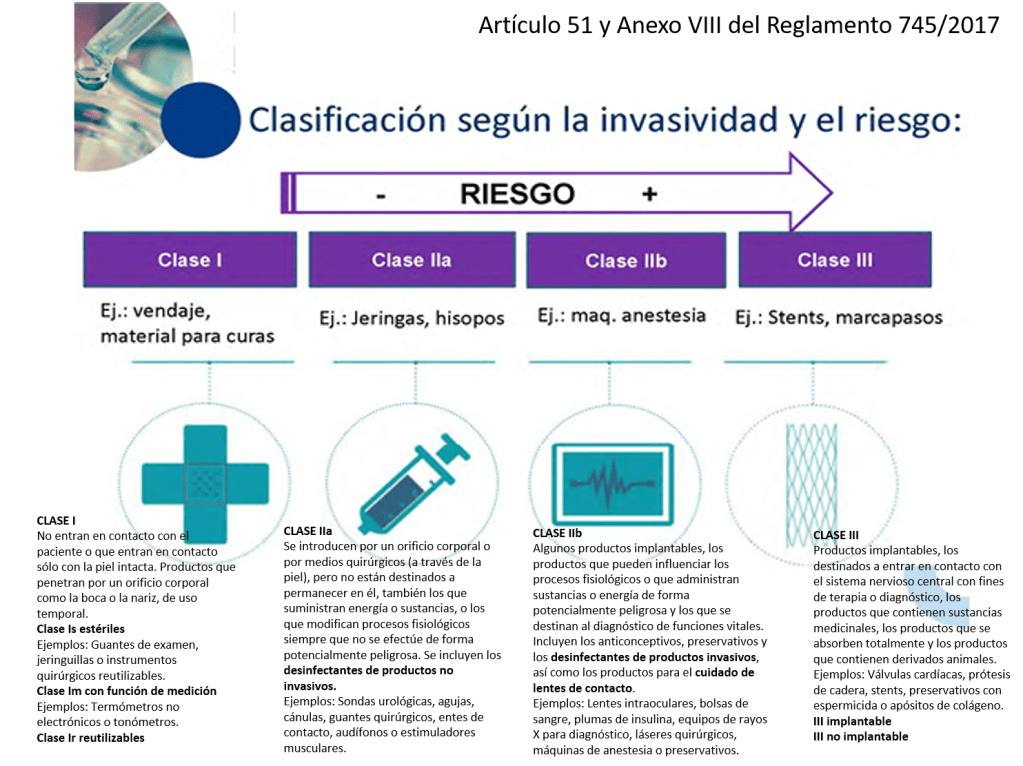
¿Cómo se clasifican los productos sanitarios? (Artículo 51 y Anexo VIII del Reglamento 745/2017)
Este reglamento tiene unos cambios relevantes que se aplican a la Central de Esterilización o RUMED:
•Los desinfectantes de productos no invasivos son IIa
•Los desinfectantes de productos invasivos son IIb
•Las lavadoras desinfectadoras pasan a ser productos sanitarios de la clase IIb
•Los equipos de esterilización pasan a ser productos sanitarios de la clase IIa
•Los indicadores químicos y biológicos NO tienen la consideración de productos sanitario (Confirmado por la AEMPS que se lo ha dicho a un amigo). La empresa GKE ha elaborado un documento donde dicen que «The GKE cleaning process monitoring indicators (CPI) are no medical devices in all countries and do not require any registration» (Párrafo actualizado 21/04/2022).
•Los productos fabricados por el propio hospital para uso interno (in house) pasan a estar regulados y precisan de documentación técnica y sistema de calidad ISO 13485 (¡¡no la 9001!!).
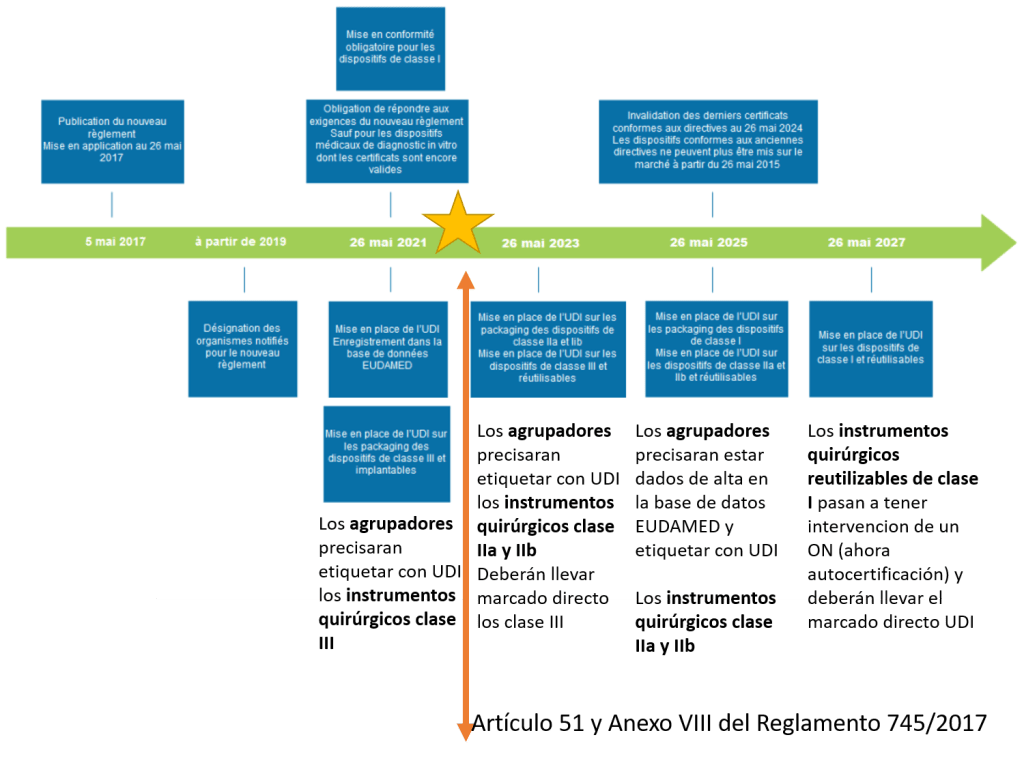
Aunque el Reglamento 2017/745 es de aplicación directa, hay determinados aspectos como el reprocesamiento, el régimen lingüístico o cuestiones de la fabricación en centros sanitarios, entre otros, que el reglamento determina que serán los Estados miembros los que establecerán la regulación a nivel nacional. Es por esto que en la actualidad se está elaborando el nuevo Real Decreto de Productos Sanitarios (sustituye al RD 1591/2009). Un producto sanitario:
-Se utiliza en personas.
–Finalidad prevista: Se diseña para cumplir un determinado fin. Documentada en el expediente técnico
-El fabricante lo ha validado en relación con el objetivo perseguido. Este objetivo lo establece el fabricante, y él mismo lo confirma, para emplearlo en relación con la enfermedad, discapacidad, proceso fisiológico, o patológico.
-Se ha diseñado para conseguir unos beneficios clínicos, siempre superiores a los posibles riesgos, y que él mismo ha sido capaz de evidenciar y justificar.
•Cuando se introduce un producto en el mercado, o lo pone en servicio, el fabricante se asegurará que se ha diseñado y fabricado con arreglo a los requisitos generales de producto sanitario.
•Realizar una evaluación clínica en relación con los requisitos esenciales de producto, incluyendo un seguimiento postcomercialización.
•Los de aquellos productos que no sean a medida, elaborarán y actualizarán la documentación técnica de dichos productos, permitiendo la evaluación de la conformidad de producto.
•Cumplir con las obligaciones de identificación y las de registro.
¿Cómo lo valida el fabricante? ¿Cómo el fabricante es capaz de determinar que, ciertamente, es capaz de ser eficaz y seguro para lo que él mismo pretendía Mediante el ejercicio de la evaluación clínica. La evaluación clínica es un proceso continuo, sistemático y planificado que pretende conseguir, evaluar y analizar información clínica. Estas normas pueden sernos de ayuda:
Y todo ello, en un sistema de seguridad y funcionamiento, que deberá disponer de un sistema de gestión de la calidad (UNE-EN ISO 13485 y la Guía o ayuda la UNE 179003:2009) y establecer, documentar, aplicar y mantener un sistema de gestión de riesgos (UNE-EN ISO 14971). Los estados miembros pueden permitir a los hospitales no aplicar todos los requisitos si la seguridad del nuevo producto son similares a las de los productos originales, el reprocesado se hace según especificaciones comunes sobre gestión de riesgos, la validación de procedimientos, liberación paramétrica o no de productos, ensayos, sistemas de calidad, etc… Solo se reprocesarán productos que se considere seguro hacerlo (por eso van a obligar a los hospitales obtener una Licencia de Funcionamiento). Los estados miembro pueden aplicar estas excepciones también a productos que se reprocesan en empresas externas a los hospital (PERO el producto vuelve al mismo hospital). Parece que los centros sanitarios y los fabricantes externos tendrán un régimen especial. Se abre la posibilidad a los «third party», aunque cada país puede imponer limitaciones al reprocesado y el uso de productos reprocesados. El reprocesamiento se lleva a cabo según especificaciones comunes sobre gestión de riesgos, validación de los procedimientos, liberación, sistema de gestión de la calidad, notificación de incidentes, trazabilidad.
En cuanto a los recursos humanos, se insiste en la figura del Responsable o Director Técnico, con unas funciones que ya conocemos:
•Supervisar las actividades de fabricación.
•Comprobar que se cumplen los requisitos exigidos por la reglamentación.
•Supervisar el archivo documental.
•Revisar y evaluar los incidentes.
•Ser interlocutor con las autoridades sanitarias y facilitar la documentación requerida.
•Solicitar la Licencia de Funcionamiento
Y aparece una nueva figura, que es el Responsable del cumplimiento del Reglamento (Artículo 15 Reglamento 745/2017), que tendrá esta formación, como Licenciado o Grado en Derecho, Medicina, Farmacia, Ingeniería u otra pertinente y un año experiencia en asuntos reglamentarios o SGC de productos sanitarios o cuatro de experiencia en asuntos reglamentarios o en SGC de productos sanitarios.
¿Y con todo esto ya vale?
Pues no, aun tenemos que (Artículo 10 del Reglamento 745/2017):
•Mantener a disposición de las Autoridades la documentación, la declaración de conformidad durante un periodo mínimo de 10 años desde la puesta en el mercado del último producto (15 años si son implantables).
•Disponer de un sistema de registro e información a las autoridades ante incidentes graves o acciones correctivas.
•A petición de la Autoridad competente, facilitarle toda la información para demostrar la conformidad de producto.
•En caso de que el fabricante encomiende a un tercero el diseño o fabricación del producto, se recogerá en la documentación.
•Las personas físicas o jurídicas podrán reclamar indemnizaciones por daños o perjuicios causados por un producto defectuoso; con arreglo al derecho nacional, o de la Unión, aplicable.
•Creación de una nueva base de datos llamada EUDAMED (acceso a las autoridades, la industria, los profesionales sanitarios y el público general) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
•Mejora en la trazabilidad de los productos sanitarios, mediante la implantación de un número de identificación único (UDI) (Artículo 27 y 28 del Reglamento 745/2017 y Anexo VI).
Para terminar unas breves reflexiones en voz alta:
Todo este Reglamento puede ser un revulsivo para las RUMED, por el papel central en la seguridad del paciente que se les da, pero ¿estarán todas preparadas? Yo creo que no. Algunos hospitales o centros pequeños no tiene personal cualificado suficiente, maquinaria y equipos validados, presupuesto para desarrollarlo…
Creo que va a haber un boom de empresas especializadas en dar cobertura a todos los hospitales. Unos dando formación y asesoría en el Reglamento, sobre todo en los aspectos normativos, sistema de gestión de la calidad y sistema de gestión de riesgos.
Y otras empresas dando servicios mediante subcontratación o externalización de la RUMED o de la Dirección Técnica y Normativa.
No sabemos cómo será el futuro real decreto pero puede cambiar toda nuestra filosofía de trabajo.
Sé que es una entrada dura y con mucha información, y con muchas dudas que tendremos que ir solucionando.
El autor no tiene conflicto de intereses con la empresa Dr. Weigert
Y la despedida con uno de los mejores temas de los 70-80, con las Baccara y su «Yes Sir, I can Boogie» que se ha convertido en el himno no oficial de la selección escocesa de futbol, donde juega el petardo de Bale. En esos años, sólo sabían inglés Jesús Hermida y el Príncipe Gitano.
Se ampliará la información de esta entrada próximamente con un análisis crítico de la Guía. Un problema informático de programación ha hecho que se publicara antes de tiempo.
Ha salido publicada esta Guía que me parece muy buena. Aquí os dejo el enlace a la misma (en francés). Los suizos además de quesos, chocolates, los caramelos Ricola y relojes hacen muy buenas Guías (¡¡y en tres idiomas!!, de los que ninguno es el inglés). Por cierto, además de las corbatas, me encantan los relojes suizos, por si os animáis a hacerme un regalo.
Por si no la conocéis, os dejo el enlace a la Guía de Transporte de material sucio a la RUMED.
Así que unimos estas Guías a la colección de las ya existentes.
La despedida será con la voz más dulce que un Ricola, un tipo más grande que el Montblanc y más movimiento que un Omega automático. Si, eso es, El Fary.
Y tan grande como José Luis Cantero es Elton John:
Seguimos con la última entrada dedicada a los endoscopios. Pero esta vez vamos a hablar de costes, algo que he tratado poco en este Blog. Aquí el último vídeo de la serie:
Cabaret (1972)
Actualmente, y por norma general, en España no se contempla que el coste del reprocesado, reparaciones, mantenimiento y control microbiológico se impute al coste total del procedimiento de endoscopia. Existen varios estudios de micro-costes que han estimado el coste por procedimiento considerando las partidas de costes atribuibles al coste capital (coste de los dispositivos, endoscopios y torres), coste de las reparaciones y mantenimiento (de los endoscopios reutilizables y los dispositivos que componen las torres de endoscopia, durante toda su vida útil), costes del reprocesado (coste del procedimiento de la puesta a punto del endoscopios reutilizable y el coste de la mano de obra que lo realiza) y el coste del control microbiológico.
Estos estudios han demostrado que el coste por procedimiento con endoscopios reutilizable es similar o incluso superior que el coste por procedimiento con endoscopios de un solo uso, debido al coste del reprocesado, del control microbiológico y de las reparaciones y mantenimiento. Hay diferentes estudios:
Este equipamiento de un solo ha venido a quedarse y su implantación es cuestión de tiempo. Se trata de una revolución en el diseño del equipamiento en endoscopia.
En lo que al coste del reprocesado se refiere, Ofstead et al (2017) llevaron a cabo una revisión pormenorizada del procedimiento a seguir para el reprocesado de un endoscopio reutilizable y determinaron todo el material necesario para llevar a cabo el reprocesado, su coste y el tiempo empleado: de este este estudio se estima que el tiempo empleado en cada reprocesado de un endoscopio es de 76 minutos y el coste total asociado al reprocesado es de 83€ por procedimiento.
La limpieza, desinfección y esterilización del broncoscopio y sus accesorios es un tema complejo, todavía no resuelto. Actualmente no existe un método de desinfección ideal en broncoscopia. Éste sería el que en un espacio de tiempo corto fuera capaz de lograr una desinfección de alto nivel sin dañar el instrumental, no fuera perjudicial para el personal que lo maneja ni para el medio ambiente y tuviera un coste económico razonable.
El tiempo empleado en el reprocesado de dispositivos reutilizables también es un recurso porque se necesita personal altamente especializado en el reprocesado y la falta de disponibilidad de un dispositivo por el hecho de que se esté reprocesando impacta en el flujo normal del trabajo, algo que con el uso de endoscopios de un solo uso no ocurriría.
Y hay otros costes como el control microbiológico. La periodicidad del control microbiológico de los endoscopios no está bien establecida en las guías nacionales, pero se pueden realizar controles cada mes y siempre que se sospeche de una posible contaminación. Del estudio de Gavaldá et al (2015) se estimó el coste asociado a la monitorización microbiológica de los broncoscopios flexibles reutilizables. Según este estudio, el coste total del test microbiológico por broncoscopio es de 111,5 euros.
La consecuencia clínica de utilizar un endoscopio reutilizable es que el paciente se infecte porque el dispositivo está contaminado. Esta consecuencia clínica tiene un coste asociado que no tendría si se utiliza un dispositivo de un solo uso. Los pacientes no se infectan porque los endoscopios de un solo uso son estériles de por sí. En el estudio de Mouritsen et al (2020) se cuantifica que el uso de endoscopios de un solo uso resulta en un ahorro de 291 £ netas (contabilizando el coste capital y reprocesado) y se evita un riesgo de infección del 2,8% usando este tipo de dispositivos. Sin embargo, los broncoscopios flexibles reutilizables conllevan un riesgo de contaminación del paciente del 15,3% lo que supone un coste adicional por procedimiento. Teniendo en cuenta el riesgo de contaminación del paciente, aScope™4 Broncho (Mouritsen) es rentable en comparación con los broncoscopios flexibles reutilizables.
Terjesen et al (2017)encuentra un ahorro de 118 US$ por procedimiento y una eliminación del riesgo de infección del 0.7% utilizando los dispositivos de un solo uso. Sin embargo, con la opción reutilizable, las estimaciones del método Delphi determinaron aproximadamente un 3% de riesgo de contaminación cruzada y aproximadamente un 21% de riesgo de infección posterior. Del análisis de coste utilidad llevado a cabo por Mærkedahl et al, (2020) se concluye que el Ambu® aScope™4 Broncho es rentable en comparación con los fibrobroncoscopios reutilizables, y se asocia con un ahorro de costes de 211,12£ y una pequeña ganancia de 0,0105 años de vida ajustados por calidad (AVAC o QALYs en inglés).
Acaba de salir en enero de 2022 este análisis en la revista Gastrointestinal Endoscopy de coste utilidad muy interesante, y en él se observa que los dispositivos de un solo uso son la opción más ventajosa (Barakat 2022).
En el Congreso de la SF2S hubo dos poster sobre el tema, aunque es difícil de valorar, por el poco espacio y no conocer toda la metodología de cálculo de costes:
El relleno de botellas de alcohol de toda la vida (garrafón), pero en versión COVID
Limpieza de endoscopios Hospital San Juan de Dios Aljarafe
La reutilización del endoscopio de un solo uso en un mismo paciente está desaconsejada ya que podría poner en peligro su salud, incluso su vida de tratarse de un paciente inmunodeprimido en muchos casos, al transmitir bacterias patógenas que colonizan los endoscopios y proliferan durante los períodos de almacenaje (McGrath 2016) (Shuman 2012).
En España sigue estando prohibida la reesterilización de productos de un solo uso. En otros lugares del mundo se está debatiendo.
Si en los endoscopios flexibles reutilizables existe la posibilidad de producir transmisiones cruzadas (Gastmeier 2013) (Kenters 2015), en un dispositivo de un solo uso, se puede incrementar la probabilidad («It is very likely that many of such outbreaks have been missed in the past because this pathogen belongs to the physiological gut flora. However, with the emergence of highly resistant (carbapenemase-producing) strains, strict adherence to infection control guidelines is more important than ever»).
En una reciente revisión narrativa (Heuvelmans 2021) sobre el tema se observa que «la eficacia de las medidas complementarias es insuficiente y que la contaminación del duodenoscopio sigue siendo un problema». Esas medidas eran «desinfección doble de alto nivel, cultivo microbiológico y cuarentena, esterilización con gas de óxido de etileno y esterilización química líquida». Se proponen alternativas como «duodenoscopios de un solo uso, agua ácida electrolizada y plasma de peróxido de hidrógeno vaporizado».
Tomado de: Heuvelmans, M., Wunderink, H.F., van der Mei, H.C. et al. A narrative review on current duodenoscope reprocessing techniques and novel developments. Antimicrob Resist Infect Control10, 171 (2021). https://doi.org/10.1186/s13756-021-01037-z
Altamente recomendable es ver la tabla 3 que se presenta en la revisión de Heuvelmans.
Si realizamos esta práctica ilegal (en España) de reesterilizar productos de un solo uso para un posible ahorro de costes, ¿debemos informar a nuestros pacientes? ¿es ética esta práctica? ¿puede llegar a ser coste efectiva?
La literatura ha tratado este tema, que no está cerrado:
Naoum 2021 («El uso repetido de SUDs en la cirugía de cataratas no es apropiado, pone en peligro la seguridad del paciente y conlleva una responsabilidad legal para el reutilizador»; «shows that it is not cost beneficial»)
Wang 2019 («La investigación indicó que, aunque la reutilización de los DUE está prohibida legalmente en China, el reprocesamiento y la reutilización en los hospitales estaban muy extendidos. La mayoría de las respuestas tendían a aceptar los SUD reprocesados si se garantizaba la seguridad y los precios bajos. Estas contradicciones existentes y la falta de investigación pertinente hicieron que los responsables políticos de China se enfrentaran a numerosos retos a la hora de construir y mejorar este sistema de uso de dispositivos médicos para satisfacer las crecientes demandas de los sectores sociales»)
Alfa 2004. Con los cambios de decisión de la FDA se ha producido un descenso de reprocesado en el propio centro y trasladando la responsabilidad a una Third-party.
Furman 2000. En EEUU existen las Third-party para el reprocesado («El reprocesamiento por parte de terceros de los dispositivos médicos etiquetados como de un solo uso es una práctica segura y regulada por la FDA que ayuda a los hospitales a reducir los costes sin comprometer la atención al paciente. El simple hecho de que un dispositivo esté etiquetado como de un solo uso no significa que no pueda ser reprocesado de forma segura. Al contrario, la etiqueta de un solo uso es elegida por el fabricante, a veces para obtener un beneficio económico, ya que no existen reglamentos o normas formales de la FDA para distinguir entre dispositivos reutilizables y de un solo uso. El actual marco normativo de la FDA para los reprocesadores de terceros, que hace hincapié en el cumplimiento de los requisitos de garantía de calidad de la FDA, está actualmente en revisión, y la agencia está en proceso de desarrollar un nuevo esquema normativo para el reprocesamiento») Estas empresas todavía no han creado un mercado en Europa.
«Bajo la estricta supervisión de la Administración de Alimentos y Medicamentos de Estados Unidos (FDA), los miembros de la Asociación de Reprocesadores de Dispositivos Médicos (AMDR) reprocesan (o limpian, prueban, reacondicionan, empaquetan y esterilizan, entre otros pasos) dispositivos médicos seleccionados etiquetados por el fabricante original como de «un solo uso». Vukelich
En junio de 2021 la FDA y la AAMI lo han establecido:
Utilizar la esterilización en lugar de la desinfección de alto nivel, cuando sea posible, «porque la esterilización tiene un mayor margen de seguridad que la desinfección de alto nivel».
Si se utiliza la desinfección de alto nivel, «los pasos de desinfección deben incluir la limpieza previa, la prueba de fugas, la limpieza, la desinfección de alto nivel, el enjuague con agua del grifo o de la red pública, seguido de un lavado con alcohol o con agua crítica (filtrada o estéril), y el secado».
Utilice únicamente los accesorios de limpieza, desinfectantes de alto nivel, agentes de limpieza enzimáticos y detergentes especificados por el fabricante. Después del reprocesamiento, almacene los broncoscopios de una manera «que minimice la probabilidad de contaminación o de acumulación y retención de humedad, de acuerdo con las instrucciones del fabricante».
Siga las recomendaciones del fabricante para el mantenimiento preventivo y la reparación del dispositivo y los accesorios.
Desarrolle programas de inspección rutinaria y mantenimiento periódico de acuerdo con las instrucciones del fabricante.
No reprocesar ni reutilizar los broncoscopios de un solo uso.
«El cambio de paradigma que se está produciendo en la asistencia sanitaria -proporcionar un servicio de la máxima calidad al menor coste asequible- ha aumentado considerablemente las expectativas de los hospitales. El sector del reprocesamiento de terceros ha sido un catalizador de dicho cambio desde su creación, y AMDR confía en que los SUD reprocesados seguirán desempeñando un papel cada vez más importante en nuestro sistema sanitario». Vukelich
El negocio del reprocesado de dispositivos médicos o productos sanitarios es un mercado al alza. Les recomiendo visitar la web de la AMDR.
Las conclusiones en general son «There is insufficient evidence to establish the safety, efficacy and cost-effectiveness of reusing SUDs. Legal and ethical issues require attention to minimize liability and maintain patient safety and trust. Some hospitals that reprocess SUDs do not have adequate documentation» (No hay pruebas suficientes para establecer la seguridad, eficacia y rentabilidad de la reutilización de los SUD. Las cuestiones legales y éticas requieren atención para minimizar la responsabilidad y mantener la seguridad y la confianza de los pacientes. Algunos hospitales que reprocesan los SUD no cuentan con la documentación adecuada) Hailey 2008
Esta historia no ha terminado y creo que se va a convertir en una historia interminable, por los intereses contrapuestos de unos y otros; y la ausencia de contabilidad analítica seria y rigurosa en el sistema sanitario (especialmente el español).
Los lectores de «cierta edad» habrán disfrutado del libro de Michael Ende y de la película «La historia interminable» o al menos su banda sonora:
Para esos más mayores. Teníamos hace unos años a Romina Power (¡qué guapa!) y Albano que con tanta «Felicitá» parecía un amor interminable, y al final todo se acabó.
Eran años donde los italianos triunfaban en el panorama musical.
El autor del Blog tiene conflicto de intereses con la empresa Ambu.
Seguimos con otra entrada dedicada a los endoscopios, después de la buena acogida de la primera.
En el Congreso de la SEMPSPH, una muy buena amiga (C.B.) de Madrid me recomendó este temazo sobre los geles antisépticos, que no debemos olvidar que son la base de la prevención.
Según el PRAN la resistencia antibiótica es desde hace años una de las amenazas más graves a las que se enfrenta la salud pública y supone uno de los retos más importantes para la medicina moderna. El aumento de la resistencia a los antibióticos se debe a diversos factores, pero el uso inapropiado e indiscriminado de estos medicamentos es uno de los que más contribuyen a la aparición de este fenómeno, que causa un gran impacto clínico, epidemiológico y microbiológico.
La resistencia antibiótica puede ser nuestra próxima pandemia. No lo digo yo, lo dijo Fernando Simón en el Congreso de la SEMPSPH en Santander (tenéis la ponencia completa en el vídeo). Y comentó que en salud pública no trabajamos para la enfermedad sino para la salud, por lo que las resistencias se deben realmente a un uso inadecuado de los antibióticos; y lo que se debe hacer es concienciar a todos los usuarios de las repercusiones de tomas de decisiones en la práctica clínica.
Los endoscopios reutilizables contaminados se han relacionado con más infecciones y brotes que cualquier otro dispositivo médico reutilizable (Rutala et al., 2016). Existen varios estudios que corroboran la contaminación de endoscopios, incluso tras su reprocesado. El uso de estos dispositivos contaminados puede llevar a que se produzcan brotes y que otros pacientes se contaminen produciéndose lo que se denomina contaminación cruzada, cuya consecuencia para los pacientes es el desarrollo de una infección cruzada. Hay numerosos casos reportados en la literatura que describen infecciones por la utilización de endoscopios contaminados, tras diferentes tipos de procedimientos, duodenoscopia, cistoscopia, broncoscopia, etc; y muchos de ellos describen infecciones por microorganismos multirresistentes (MDRO). Los endoscopios son los dispositivos médicos que se asocian con mayor frecuencia a los brotes de infecciones nosocomiales. Los microorganismos resistentes emergentes suponen una preocupación cada vez mayor para la población y para los responsables del control de las infecciones. Hay gran variedad de microorganismos patógenos, cuyas fuentes de infección son principalmente los materiales semicríticos como los endoscopios. La contaminación de los endoscopios ya reprocesados y listos para su uso se ha vinculado a infecciones de pacientes, incluidas infecciones por bacterias multirresistentes a medicamentos, de ahí que algunas guías propongan la profilaxis en algunas intervenciones y en algunos pacientes.
No existe el riesgo 0 de transmisión de infecciones graves a través de endoscopios reprocesados. En el Congreso de la SEMPSPGS de Santander, la Dra. Ana Haro expuso la importancia de la desinfección de alto nivel (DAN) en diversos materiales.
Broncoscopio (Mehta & Muscarella 2019) (Ofstead et al., 2018) (Gavaldá et al., 2018) (Kovaleva et al., 2013) (Mouritsen et al., 2019). Los broncoscopios pueden suponer un riesgo potencial y poco reconocido de transmisión de Enterobacterias Resistentes a Carbapenem (CRE) y MDROs. Así se puso de manifiesto en una revisión de la literatura llevada a cabo por Mehta y Muscarella 2019 , cuyo objetivo principal fue investigar el riesgo de transmisión de infecciones por CRE y MDROs asociado a los broncoscopios, así como, evaluar si las medidas extra serían útiles y aconsejables para mejorar la seguridad y la eficacia del reprocesado de los broncoscopios. Ante la posibilidad de contaminación e infecciones cruzadas y la aparición de complicaciones posteriores, algunas guías de práctica clínica (SEPAR) proponen la profilaxis antibiótica previa a los procedimientos de endoscopia en perfiles de pacientes determinados como los grandes inmunodeprimidos ante una maniobra como una broncoscopia.
En los últimos años, la FDA ha publicado comunicaciones relacionadas con la seguridad de los endoscopios reutilizables y la potencialmente comprometida seguridad del paciente. Dado que la esterilización no eliminará las infecciones cruzadas asociadas a los broncoscopios, y contribuirá a la prolongación del tiempo de respuesta, al aumento de las tasas de reparación, a la disminución de la capacidad asistencial y al aumento de los costes, la solución es la implementación de los broncoscopios de un solo uso. Deberían introducirse para todos los procedimientos en la UCI, y además para todos los pacientes con infecciones previas por MDRO, pacientes inmunocomprometidos, pacientes con COVID-19, o pacientes con enfermedades prionicas, y también cuando no sea posible reprocesar los dispositivos de inmediato. Al implantar el uso de broncoscopios de un solo uso para todos los pacientes colonizados por MDRO, el riesgo de infección cruzada por broncoscopio se reduce entre un 0,9% y un 2,8% según la recomendación de la FDA. Por consiguiente, al disminuirse el riesgo de infecciones por MDRO, el ahorro de costes asciende a 186-812$ por broncoscopia. Además, si se considera que todos los pacientes de la UCI están inmunocomprometidos, el ahorro de costes al implantar la broncoscopia de un solo uso para estos pacientes oscilará entre 227$ y 703$ por broncoscopia.
Y después de tanta contaminación, cerramos la entrada con música relacionada. «Contamíname» de Pedro Guerra:
Ya sabéis que me gusta más la música freaky y movidita. Este temazo de los «Coches de choque» es inolvidable para todos. Si a alguien no le produce recuerdos, es que no ha ido nunca a una feria o fiesta de pueblo. Y es que hace poco una persona me oyó tocar con la trompeta un pasodoble muy conocido, y me dijo que nunca había bailado un pasodoble en la plaza de un pueblo (¡¡se me cayó el mundo!!).
El autor del Blog tiene conflicto de intereses con la empresa Ambu.
Vuelvo a tratar en este blog el tema de los endoscopios por su importancia en el sistema sanitario, y porque cada vez se han vuelto técnicas más invasivas y comunes. Hace poco se publicó la Guía AKI.
Se trata de cuatro entradas que he elaborado con la empresa Ambu. Cada una de estas cuatro entradas se acompañan de un vídeo sobre el tema de la misma.
Los endoscopios flexibles son productos sanitarios semicríticos que requieren una limpieza en profundidad si se van a reutilizar. Para la reutilización es necesario seguir un proceso de desinfección o esterilización. Se define la esterilización como la destrucción de todos los microorganismos viables, presentes en un objeto, incluidas las esporas bacterianas. Por otro lado, la desinfección es el procedimiento que destruye, en mayor o menor grado, las formas de microorganismos metabólicamente activas, aunque no las esporas bacterianas. Por tanto, para paliar el riesgo de infección cruzada, se recomienda introducir un paso de esterilización durante el reprocesado de los endoscopios flexibles reutilizables.
Actualmente, los endoscopios se consideran dispositivos semicríticos según el sistema de clasificación Spaulding, ya que están en contacto con las membranas mucosas. El sistema Spaulding clasifica los dispositivos médicos en tres categorías (no críticos, semicríticos y críticos) en función del riesgo de infección. Los dispositivos clasificados como semicríticos requieren una desinfección de alto nivel (DAN). Las directrices de reprocesamiento actuales recomiendan más de 100 pasos para el reprocesado de cada endoscopio y, aunque existen similitudes entre las directrices, algunas recomendaciones no son universales. Debido al riesgo de infecciones cruzadas entre pacientes provocadas por endoscopios gastrointestinales contaminados, se ha sugerido que los endoscopios gastrointestinales deben clasificarse como dispositivos críticos en lugar de dispositivos semicríticos, lo que requeriría que los endoscopios se sometieran a una esterilización a baja temperatura. Dado que los endoscopios gastrointestinales son termolábiles, solo es posible utilizar la DAN con agentes químicos o tecnologías de esterilización a baja temperatura (Rutala et al., 2016).
El riesgo es inherente a la intervención en endoscopia, siendo mayor en la endoscopia terapéutica, con rotura de las barreras anatómicas.
Sin embargo, la evidencia reciente demuestra los principales desafíos asociados con la esterilización. Un metaanálisis llevado a cabo por Larsen et al., (2020) demostró que el riesgo del 15% de contaminación cruzada de endoscopios después de la Desinfección de Alto Nivel (DAN) sólo se redujo al 9% después de añadir un paso de esterilización. Incluso la doble DAN y la esterilización no lograron disminuir significativamente la tasa de contaminación. Esto demuestra claramente que la adición de un paso de esterilización probablemente no resolverá los problemas de contaminación cruzada por sí sola.
Al mismo tiempo, existe la percepción de que el uso de endoscopios reutilizables tiene un menor impacto económico y ambiental si se compara con el uso de endoscopios de un solo uso. Sin embargo, hay varios estudios que muestran un impacto medioambiental similar entre los endoscopios de un solo uso y el reprocesado de endoscopios, principalmente debido a los métodos de reprocesado.
Snyder et al., (2017) compararon la frecuencia de contaminación de los duodenoscopios con organismos multirresistentes (MDRO) o cualquier otra bacteria después de la desinfección o esterilización siguiendo 3 métodos diferentes. Hallaron que los métodos de desinfección y esterilización (doble DAN o DAN/EtO) no proporcionaron una protección adicional contra la contaminación, se observó crecimiento bacteriano superior a 0 UFC en 16,1% de los duodenoscopios del grupo de DAN, 16,0% en el grupo de doble DAN y 22,5% en el grupo de DAN/EtO. Larsen et al. (2020) determinaron una ratio de contaminación del 9,20% tras doble DAN/EtO y un 16.14% tras HLD (doble alta desinfección). La ratio de contaminación de duodenoscopios reprocesados era del 15,25%. Cabe resaltar también que la JCI (Joint Comission International) en este artículo técnico clasifica los endoscopios flexibles, los gastroscopios y los broncoscopios como los dispositivos más difíciles de limpiar y esterilizar (ver su tabla 1), por ser dispositivos complejos en su diseño y ensamblaje, lo que no permite asegurar que tras reprocesarse estos queden suficientemente limpios o estériles, ni que funcionen correctamente, suponiendo todo ello un riesgo para el paciente.
¿Y si un doble de Barry White te hiciera una colonoscopia con mucho cariño?
Repercusiones medioambientales del reprocesado de endoscopios
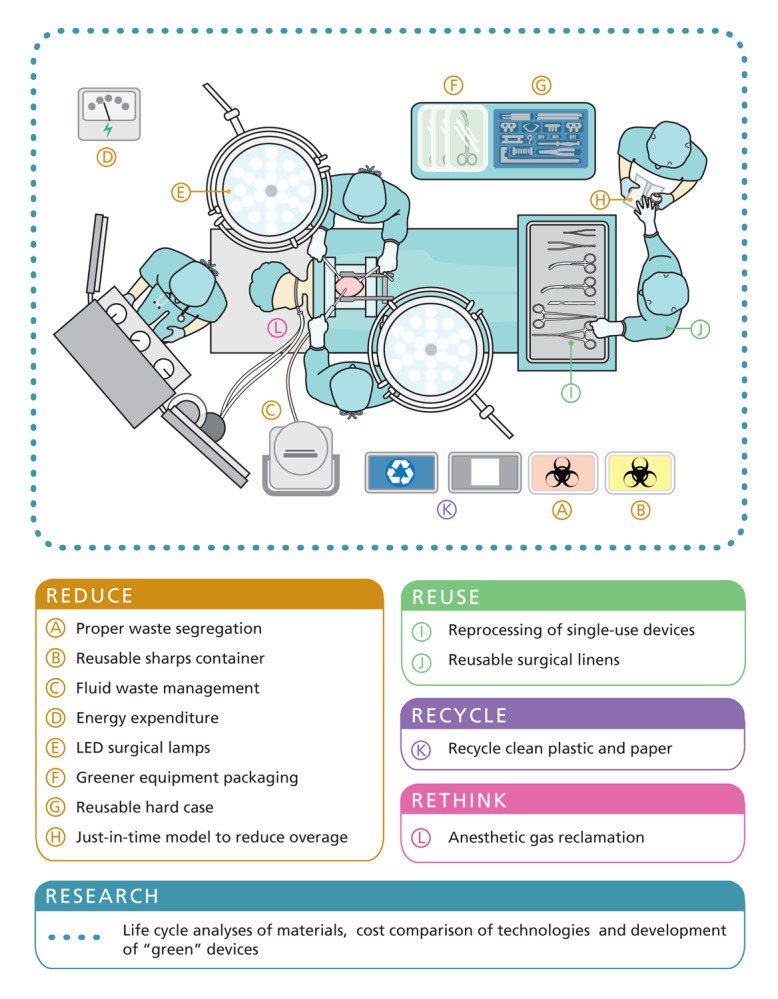
Cada vez dedicamos más recursos al sistema sanitario, y a su vez la población conocedora de los avances médicos, tiene una menor tolerancia. Para intentar conseguir un equilibrio, cada vez más necesitamos la implicación de los profesionales. Cada día hablamos más de Economía circular.
La producción y el mantenimiento de los equipos médicos puede tener un impacto medioambiental significativo y dejar una gran huella de carbono, independientemente de si son reutilizables o de un solo uso. Sin embargo, son pocos los estudios publicados hasta el momento que documentan este impacto. En particular, en un estudio llevado a cabo en 2018 por Davis et., (2018), se comparó el impacto medioambiental de los ureteroscopios de un solo uso frente a los reutilizables; el estudio determinó que el impacto medioambiental era comparable, produciendose 4,43kg y 4,47kg de CO2, respectivamente.
Otro estudio, como el de Wong et al. (2021) , también demostraron que, debido al uso de detergentes y equipo de protección individual, los endoscopios reutilizables se asociaron con iguales o mayores emisiones de equivalentes de CO2 y consumo de recursos que los endoscopios de un solo uso. En el estudio Sorensen et al., (2018) determinaron que los endoscopios reutilizables se asocian a un consumo de materiales y energía comparable o incluso mayor que los endoscopios de un solo uso, así como mayores emisiones de equivalentes de CO2, debido a que los endoscopios reutilizables deben someterse a un complejo y exhaustivo reprocesado entre usos.
Ya hice una entrada de VPRO® versus Sterrad®, pero esta entrada es al revés, es la de Sterrad® versus VPRO® (parece lo mismo pero no lo es). En aquella entrada, la empresa Steris se comparaba con ASP; pero ahora es ASP que se compara con Steris, de ahí las matizaciones. Y como siempre digo, si dudamos utilicemos fuente de información fiables. Y me encanta lo que dicen sobre los controles de calidad (frecuencias de indicadores, controles…) que dependerá de lo que decida el técnico o encargado de la central.
Si lo hice antes, lo debo hacer ahora con esta comparativa y ofreceros la tabla que nos dan. Es como el lema de los Reyes Católicos «Tanto monta, monta tanto, tanto Isabel como Fernando» (y este no es el logo del antiguo régimen español).
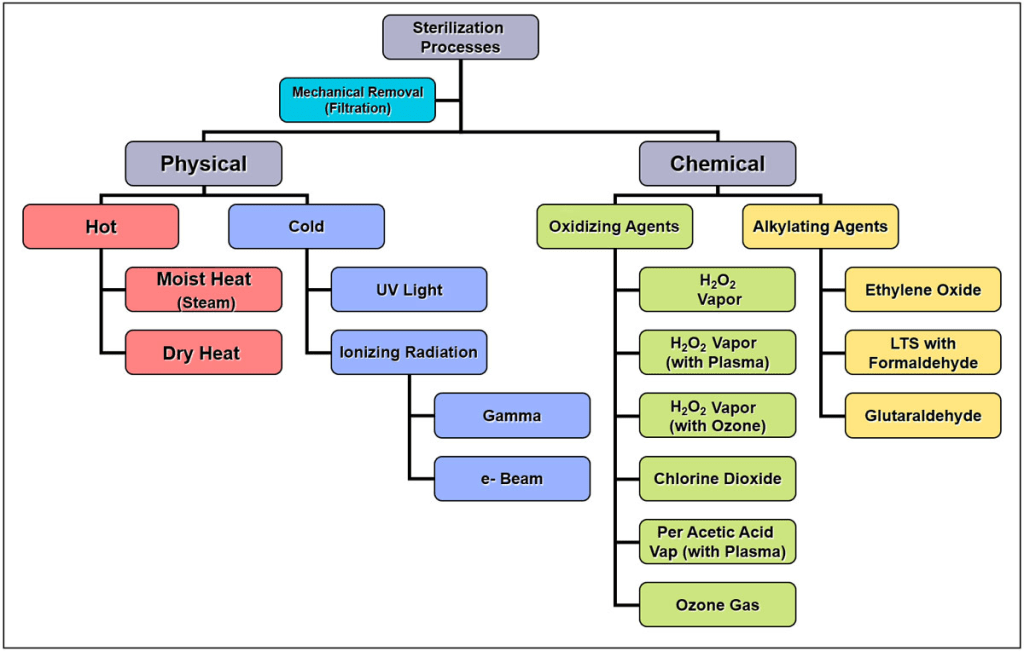
El peróxido de hidrógeno en sus diferentes presentaciones es uno más de los llamados sistemas de esterilización en frío mediante agentes oxidativos, y está pegando fuerte.
Según Steris, el VPRO gana a Sterrad en rendimiento y compatibilidad de dispositivos médicos. Veamos lo que dice ASP frente a Steris.
Debe quedar claro que se compara el Sterrad 100NX frente al VPROmax2. Se dice que el Sterrad 100NX tiene un volumen de 152 litros frente a los 136 litros del Vpromax2; pero todos sabemos que volumen no es sinónimo de capacidad. Y el Sterrad 100 NX tiene una capacidad de 93,4 litros, en el caso del Vpromax1 es de 136 litros (atención es el 1 no el 2). Hay un gráfico con unas medidas en centímetros, y lo cálculos que me salen para el Sterrad 100NX es de 157 litros, y para el Vpromax2 de 135 litros (es el tamaño de la cámara). El Sterrad 100NX es 9,5 cm más ancho, lo que podría favorecer la carga de más dispositivos para esterilizar.
Existe una diferencia entre equipos, el Sterrad utiliza el gas plasma y el VPRO una forma vaporizada de peróxido de hidrógeno. También hacen referencia a un estudio propio de ASP (la número 4) «En un estudio comparativo, STERIS V-PRO® maX demostró que las mediciones pico instantáneas de H2O2 llegaron hasta 20 ppm en la zona de respiración del usuario, por encima de los límites permisibles de ACGIH® (5 ppm). Los sistemas STERRAD™ nunca superaron 0,3 ppm» (es un estudio propio, no publicado en un revista con revisión por pares). Comentan que como ventaja del Sterrad no hay que «comprobar semanalmente la presencia de fugas en la cámara y sin necesidad inspeccionar las juntas trimestralmente». Yo soy muy de hacer pruebas, comprobaciones y checklist (la presión de las ruedas de mi coche la miro mensualmente), por tanto, lo de revisar semanal y trimestralmente puede que me deje más tranquilo con un equipo (es una opinión personal).
Presentan algunas diferencias en cuanto a los indicadores y el uso de dispositivos de desafío de proceso, que me parece interesante tener en cuenta, especialmente en material canulado como endoscopias, donde cada vez más estamos exigiendo esterilización. Conviene recordar el tipo de indicadores que existen:
Os dejo unos artículos sobre el tema de los indicadores, donde comparan varios tipos de indicadores, y que merecen la pena (artículo 1, articulo 2, articulo 3 y uno de los artículos de Kirk que es de pago, también podéis ver la tabla de la página 19 de este documento, y así os queda más claro). Y es que los indicadores dependen de varios factores, tal y como viene en este gráfico descrito. Debemos tener cuidado con su colocación en la cámara, el viraje del color, son muchos aspectos a tener en cuenta.
También se ofrece una comparativa de ciclos y tiempos; ahí estamos nosotros para valorar la penetración en equipos canulados y su uso en dispositivos de endoscopia en general. Por tanto, antes de elegir un equipamiento para nuestro centro, siempre deberemos saber la compatibilidad de ese equipamiento con nuestro instrumental y dispositivos, rentabilidad y rotación que vamos a dar, y la flexibilidad en la gestión.
Pese a que son anglosajones y no les importa hablar de dinero, no se menciona el precio de los equipos, consumibles, precio de la instalación, un SAT o servicio de asistencia técnica permanente. El coste económico es algo que siempre deberemos valorar.
¿Y los priones? Nadie habla de ellos. Se les habrá olvidado.
En el Congreso de la SF2S hubo tres ponencias sobre el peróxido, que creo que os pueden interesar:
En este Blog he procurado hablar de todos los equipamientos existentes sobre el peróxido de hidrógeno, así que será el usuario final el que debe elegir y valorar qué es lo que le viene bien:
Como no tengo conflicto de intereses con ninguna casa comercial, me permito el lujo de presentar todos los equipos sin diferencias ni sesgos comerciales.
Si queréis seguir comparando poneros a hacerlo con C. Tangana, La Húngara y El Niño de Elche. Y a mi que me recuerdan a Camela en todo.
Y aquí Camela (en rama); ya me imagino conduciendo un cochecito de choque en la ferias de Talavera de la Reina (pronto esa musiquita cañera).
Según la Resolución de 2 de junio de 2021 de la AEMPS, los antisépticos destinados al campo quirúrgico preoperatorio y a la desinfección del punto de inyección serán considerados medicamentos en lugar de biocidas. Se establece un plazo para la adaptación de los productos y las instalaciones de fabricación hasta el 1 de junio de 2022. Ya había hablado sobre la clasificación de desinfectantes,biocidas. Y se sabe que sirven para el Satisfayer® femenino y masculino.
SOLO serán los antisépticos destinados al campo quirúrgico preoperatorio y a la desinfección del punto de inyección. No se incluyen por ejemplo las soluciones hidroalcohólicas.
Los antisépticos destinados al campo quirúrgico preoperatorio y a la desinfección del punto de inyección, hasta ahora, han tenido la consideración de biocidas para piel sana, tal y como se indicaba en la Nota informativa de la AEMPS. Pero, teniendo en cuenta la tendencia europea actual de considerar los productos con esta finalidad como medicamentos, y en respuesta a las peticiones de los foros de pacientes y profesionales sanitarios, la AEMPS ha modificado la regulación en este aspecto.
De acuerdo con la reglamentación aplicable, estos productos deberán presentar una solicitud de autorización de comercialización como medicamentos. Además, el dossier de registro deberá cumplir con lo establecido en la normativa europea de referencia, así como con lo establecido en el Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, y, más concretamente, en el Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. La elección de la base legal es una decisión del solicitante, en función de los datos disponibles en la solicitud. Sin embargo, al ya existir medicamentos autorizados de estas características, se recomienda, siempre que sea posible, y con el fin de facilitar esta transición, que en caso de existir un medicamento de referencia se solicite un dossier híbrido. Las diferencias respecto al medicamento de referencia deben ser adecuadamente justificadas, en caso contrario, el expediente de registro deberá ser un expediente completo, pudiendo en este caso ser un uso bien establecido.
Las empresas que lleven a cabo la fabricación industrial de medicamentos, incluyendo el control de calidad, deberán cumplir con lo dispuesto en el Real Decreto 824/2010, de 25 de junio, por el que se regulan los laboratorios farmacéuticos, los fabricantes de principios activos de uso farmacéutico y el comercio exterior de medicamentos y medicamentos en investigación. En particular, están obligados a solicitar la autorización correspondiente, aportando para ello la información requerida en el artículo 7 del citado real decreto. El procedimiento incluye la realización de una inspección previa a la emisión de la autorización.
Biocidas
Tienen esta consideración los antisépticos para piel sana y los desinfectantes de ambientes y superficies utilizados en los ámbitos clínicos o quirúrgicos y que no entran en contacto con el paciente directamente, tales como los destinados a pasillos, zonas de hospitalización, zonas de atención y tratamiento, mobiliario, etc.
Real Decreto 1054/2002, de 11 de octubre
Medicamentos
Tienen esta consideración los desinfectantes que se destinan a aplicarse en piel dañada: heridas, cicatrices, quemaduras, infecciones de la piel, etc., así como los destinados al campo quirúrgico preoperatorio (excepto los antisépticos de piel sana destinados al lavado quirúrgico de manos), y los destinados a la desinfección y mantenimiento del punto de inyección.
Real Decreto 1345/2007, de 11 de octubre
Producto sanitario
Tienen esta consideración los productos que se destinan específicamente a la desinfección de productos sanitarios.
Real Decreto 1591/2009, de 16 de octubre Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017
a) Desinfectantes de productos sanitarios no invasivos: desinfectantes destinados a incubadoras, camillas, monitores, etc. Se clasifican como clase IIa. b) Desinfectantes de productos sanitarios invasivos: desinfectantes destinados a endoscopios, instrumental quirúrgico, etc. Se clasifican como clase IIb.
(Elaboración propia)
Si algún lector tiene tiempo, le propongo este artículo de divulgación (creo que en el mundo sanitario lo llaman transferencia, y luego cobran sexenios). Apareció el pasado domingo (14-10-2021) en «The Conversation» con el título ¿Qué limpia mejor, el jabón, los desinfectantes o los sanitizantes?, que nos habla de los primeros documentos donde aparece el uso del jabón. El primer uso documentado de jabón está descrito en una tableta cuneiforme elaborada hace 4 500 años y encontrada en la antigua ciudad sumeria de Girsu, en lo que hoy es el sur de Irak. «Ya los babilonios usaban el jabón…»
Leer legislación es muy aburrido y tedioso, pero hay que hacerlo para estar al día. Para pasar el aburrimiento, un poco de música movida. El clásico Stacy’s Mon en versión años 30. Han pasado los años y me sigue gustando la mamá de Stacy.
Me dicen mis hijos adolescentes, que esto es más trending, aunque los modelos son más de los años 70 tipo «Cuéntame».


















































Debe estar conectado para enviar un comentario.